The microfluidic 3D cell culture platform consists of 40 perfused microfluidic units (Figure 1a,b), which is used to study angiogenic sprouting of perfused microvessels against a patterned collagen-1 gel (Figure 1c). These microvessels are continuously perfused and exposed to a gradient of angiogenic growth factors (Figure 3a-d). The angiogenic sprouts can be either studied 2 days after gradient exposure or cultured for more than 5 days after gradient exposure to study anastomosis and sprout stabilization (see timeline, Figure 1d).
Seeding the iPSC-ECs using the passive pumping method should result in homogenous seeding densities (Figure 4a,b). Culture under continuous perfusion resulted in confluent microvessels in 2 days, with the cells completely lining the circumference of the microfluidic channel and the formation of a confluent monolayer against the patterned collagen-1 gel.
Exposure to a gradient of angiogenic factors resulted in directional angiogenic sprouting of the microvessels within the patterned collagen-1 gel (Figure 5a-g). Clear tip cell formation and invasion into the collagen-1 gel was visible 24 h after addition of the angiogenic gradient, while stalk cells including lumen formation were visible after 48 h (Figure 5a).
After fixation and staining, the capillary network can be visualized using phalloidin to stain F-actin and using Hoechst 33342 to stain the nucleus (Figure 5b,c). These sprouts can be quantified (e.g., shape and length14). Without addition of growth factors, no invasion into the collagen-1 gel should be observed (Figure 5d). Confocal imaging was used to determine the sprout diameter and to confirm lumen formation (Figure 5e-g).
The sprouts continue to grow towards the direction of the gradient and reach the opposite perfusion channel within 3−4 days after addition of angiogenic growth factors. This results in remodeling of the vascular network, with a clear reduction in the number of angiogenic sprouts (Figure 6a). Lumen formation was assessed by perfusion of the vascular network with fluorescently labeled macromolecules (e.g., albumin or dextrans). Perfusing the microvessels with 0.5 mg/mL labeled albumin before and after anastomosis revealed a clear difference in sprout permeability after 10 min (Figure 6b-e), which suggests that the capillaries stabilize and mature after anastomosis.
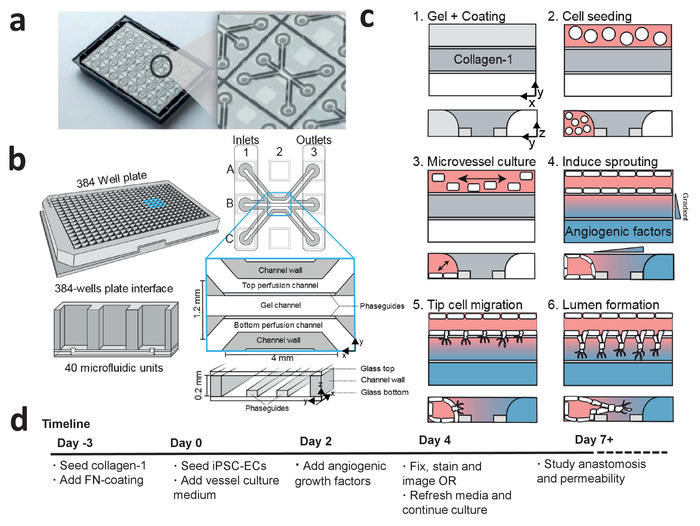
Figure 1: Microfluidic cell culture protocol for iPSC-derived microvessels. (a) The bottom of the microfluidic cell culture device is shown displaying the 40 microfluidic units that are integrated underneath the 384-well plate. Larger view displays one of the 40 microfluidic units. (b) Each microfluidic unit is positioned underneath 9 wells with 3 inlet wells and 3 outlet wells. The microfluidic channels are separated by ridges (‘phaseguides’), which enable the patterning of hydrogels in the central channel (‘gel channel’) while there is still contact with the adjacent channels (‘perfusion channels’). (c) Method to culture a perfused microvessel within the microfluidic device, which is used to study gradient driven angiogenic sprouting through a patterned collagen-1 matrix. (d) Timeline for studying angiogenic sprouting and/or anastomosis. This figure has been modified from van Duinen et al.14. Please click here to view a larger version of this figure.
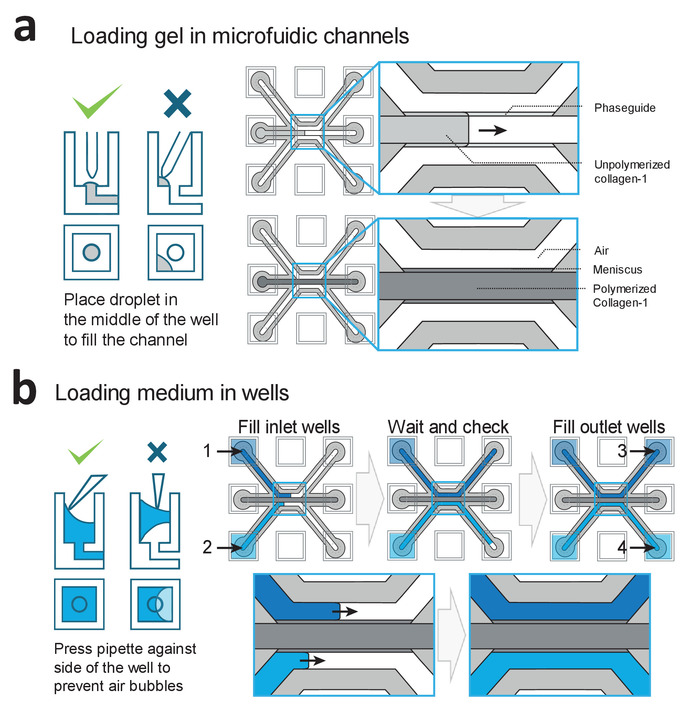
Figure 2: Loading procedures for gel and medium. (a) Examples of correct and incorrect gel deposition. Correct filing results in a patterned collagen-1 gel in the middle channel, which is subsequently polymerized. (b) Examples of correct and incorrect filling of the wells. Wells are filled in the order of 1−4 to prevent air-bubble trapping within the microfluidic channels. Please click here to view a larger version of this figure.
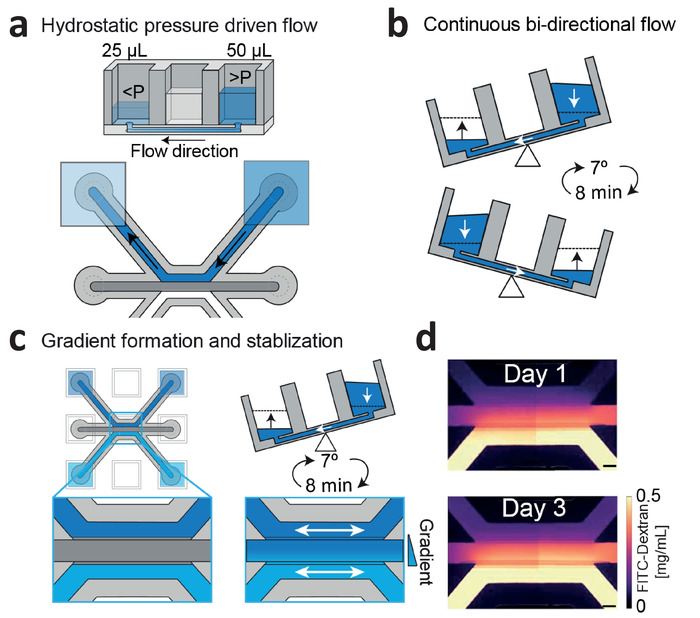
Figure 3: Continuous hydrostatic pressure driven flow and gradient stabilization. (a) Hydrostatic pressure differences between wells result in passive levelling and flow within the microfluidic channels. (b) Placing the device on a rocker platform set at 7° and 8 min cycle time results in continuous, bi-directional perfusion within the microfluidic channels. (c) Gradients are formed by introducing two different concentrations within the wells, which are continuously refreshed by passive leveling. (d) Gradient visualization using fluorescein isothiocyanate (FITC)-dextran. Bi-directional flow stabilizes the gradient up till 3 days. Scale bar = 200 µm. This figure has been modified from van Duinen et al.14. Please click here to view a larger version of this figure.
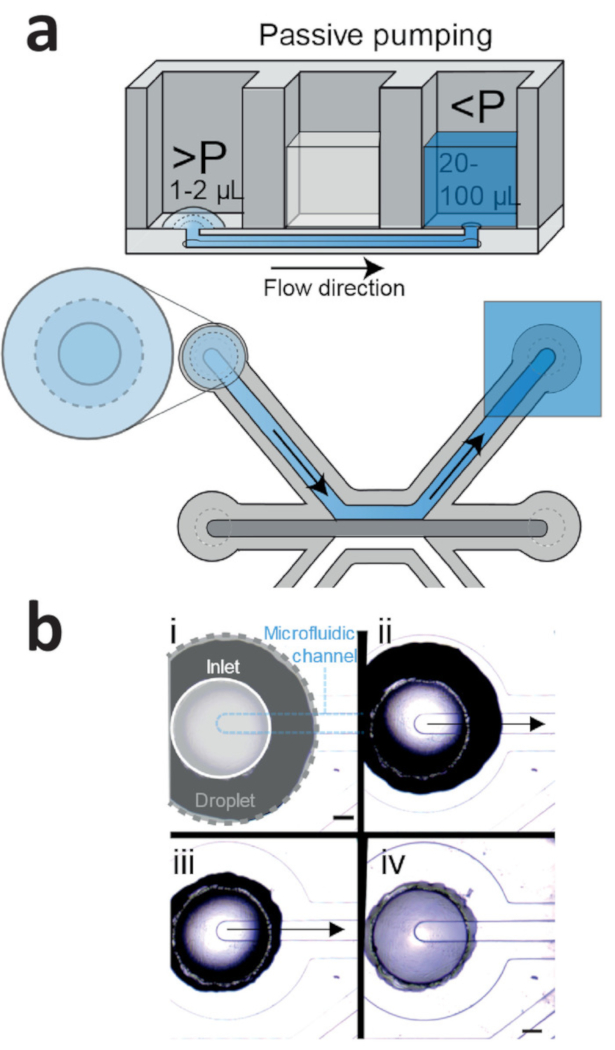
Figure 4: Passive pumping method for cell seeding. (a) Passive pumping is driven by pressure differences that are caused by differences in surface tension. This results in a flow from the droplet (high internal pressure) towards the reservoir (low internal pressure). (b) Time lapse of a droplet (gray outline) that is placed on top of the inlet (white outline) of the microfluidic channel (blue outline). Right after addition (Figure 4b, i), the droplet on top of the inlet shrinks (Figure 4b, ii: 1 s after addition; iv: 2 s after addition), which results in a flow towards the outlet. This continues until the droplet meniscus is pinned by the inlet (Figure 4b, iv). Scale bar = 400 µm. Please click here to view a larger version of this figure.
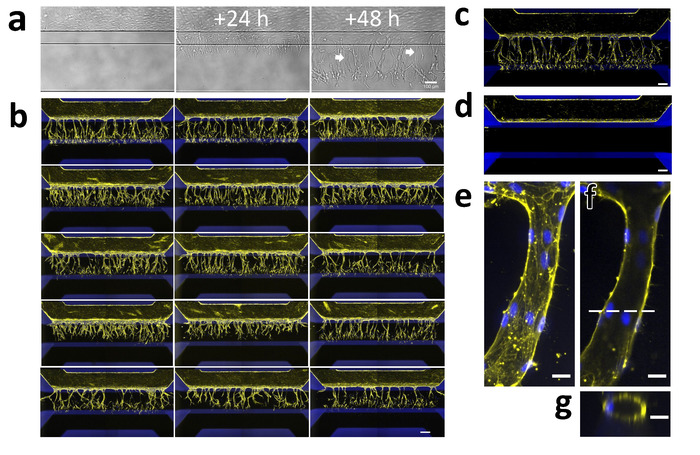
Figure 5: Robust 3D sprouting of iPSC-EC microvessels. (a) Sprouting of iPSC-EC over time. Microvessels were grown for 48 h (right) and then stimulated with an angiogenic cocktail containing 50 ng/mL VEGF, 500 nM S1P, and 2 ng/mL PMA. The first tip-cells that invade the collagen-1 scaffold (middle) are visible 24 h after exposure. The first lumens are visible (arrows) 48 h after exposure (right) while the tip-cells have migrated further in the direction of the gradient. (b) Array of 15 microvessels that were stimulated with VEGF, S1P and PMA for 2 days and stained for F-actin (yellow) and nuclei (blue). Scale bar = 200 µm. (c) Stimulated microvessel (positive control). (d) Unstimulated microvessel (negative control). (e) The maximum projection of a single capillary within the gel. (f) Same as (g) but focused on the middle. Dotted line indicates the position of the orthogonal view in panel g. Scale bars (a-d: 200 µm; e-g: 20 µm). Please click here to view a larger version of this figure.

Figure 6: Visualization of angiogenic sprout permeability before and after anastomosis. (a) Anastomosis with basal channel triggers pruning and maturation of angiogenic sprouts. Closeup of capillary bed at 2, 4, 6 and 7 days after stimulation with angiogenic growth factors. (b) Angiogenic sprouts after 2 days after addition of angiogenic growth factors. Angiogenic sprouts are formed within gel, but are not yet connected to the bottom perfusion channel. (c) Perfusion of the microvessel with 0.5 mg/mL albumin-Alexa 555 solution. Fluorescent images obtained at 0 and 10 min. (d,e) Same as in panels b and c, but after 7 days of stimulation. Sprouts are connected to the other side and formed a confluent microvessel in the basal perfusion channel. Scale bars = 100 µm. Please click here to view a larger version of this figure.
| Problem | Cause | Solution |
| Collagen-1 does not enter or fill the channel completely | Collagen-1 droplet is not placed on top of the inlet | Carefully place the droplet on top of the inlet from the gel channel |
| Volume of collagen-1 is too low | Use 1.5 µL of the gel to fill the channel completely | |
| Collagen-1 is too viscous | Use another batch of collagen-1 | |
| Collagen-1 flows into perfusion channels | Collagen-1 is pipetted directly into the inlet of gel channel | Carefully place the droplet on top of the inlet from the gel channel |
| Collagen-1 is not clear/fiber formation | Collagen-1 is not stored properly | Store collagen-1 at 4 °C, do not freeze |
| NaHCO3 and HEPES are not mixed well before adding collagen-1 | Carefully mix the NaHCO3 and HEPES by pipetting before adding the collagen-1 | |
| Droplet does not shrink using passive pumping method | Droplet adheres to side of the well | Aspirate droplet and add new droplet on top of the inlet |
| Make sure the outlet well is filled with at least 20 µL of medium | ||
| No sprouting is observed | Growth factors are not added or aliquots are not stored properly | Prepare fresh angiogenic sprouting medium |
| Air bubble blocks perfusion/ gradient formation | Remove air bubbles using a P20 or P200 pipette | |
| Volume differences between wells | Volumes in all wells need to be equal in order to form a linear gradient | |
| Cells not viable | Plate not placed on rocker platform/rocker platform turned off | Make sure rocker platform is on and has the right cycle time/angle (8 min/7°) |
| No perfusion possible due to presence of air bubbles | Remove air bubbles using a P20 or P200 pipette | |
| No lumens are formed, cells migrate as single cells | Angiogenic sprouting mixture was added before a monolayer was formed | Wait an additional 24 h before adding the angiogenic growth factors |
| Major variation in sprouting density | Differences in cell densities after seeding | Check if cell density is homogenous and comparable between microfluidic units. Add another droplet of cell suspension if necessary |
Table 1: Troubleshooting common errors.
