Purification of the α– and β-subunits of the tryptophan synthase
The α-subunit (αStTS) and the β-subunit (βStTS) of the Salmonella typhimurium tryptophan synthase were subcloned in the modified pET SUMO vector. Figure 1A shows representative SDS-PAGE results of two strong bands corresponding to the His6-SUMO-αStTS (lane αON) and His6-SUMO-βStTS (lane βON) fusion protein. The purification protocol described in this work allowed purification of both subunits individually within 2 days. The first day was used to purify each protein by Ni-NTA affinity chromatography, ammonium sulfate precipitation followed by His-SUMO-tag cleavage, removal of His-SUMO-tag traces, and protein concentration. Figure 1B and 1C show representative SDS-PAGE results of the α-subunit and the β-subunit purification, respectively. On the second day, the concentrate α-subunit, β-subunit, and the α2β2 StTS complex from the α- and β-subunits were loaded on a size exclusion chromatography column. Figure 1D shows a typical elution profile of αStTS, βStTS, and α2β2StTS complexes on a S-200 HR size exclusion column. Figure 1E shows a representative SDS-PAGE result of the collected peak fractions. The purest peak fractions were pooled, concentrated, and the α2β2 StTS complex was used for protein crystallization studies.
Purification of the wild type and mutant α2β2 StTS complex
Another rapid and efficient protocol to purify the wild type and mutant form of the α2β2 S. typhimurium tryptophan synthase complex is described in this work. Figure 2 shows a representation of the pEBA-10 construct containing the wild type translationally coupling gene (trpA and trpB) encoding for the α– and β-subunits22. The two-step PCR mutagenesis protocol to generate mutant forms of the α2β2 StTS complex is depicted in Figure 3.
The coding regions of mutant α2β2 StTS complex in pEBA10 were confirmed by DNA sequencing and used to transform E. coli strain CB149 cells26. The wild type and mutant form of the α2β2 StTS complex were overexpressed and the recombinant proteins were purified successfully within 1-2 days. Ammonium sulfate fractionation at room temperature readily removed most of the contaminant proteins from the heterologous expression system (Figure 4A, lanes 20P, 30P and 40S). A representative elution profile with relative elution position of α2β2 StTS (143.06 kDa) complex on a HiPrep 16/60 Sephacryl S-200 HR size exclusion chromatography column is shown in Figure 4B. The purity of the excluded peak fractions was SDS-PAGE analyzed before pooling (Figure 4C).
Optimization of wild type and mutant α2β2 tryptophan synthase complex crystallization
Aliquots of wild type and mutant α2β2 StTScomplex at 15 mg ml-1 were used to set up 24-well sitting drop plates. Typically, droplets consisting of 5 µL protein solution and the equivalent volume of reservoir solution were equilibrated against 500 µL of reservoir solution (Figure 5). Spermine is required to crystallize the wild type and mutant α2β2 StTS complex22. While the final concentration of spermine to crystallize the wild type is 2 mM, the concentration of spermine to crystalize the mutant complex in this work showed to be slightly higher (4-8 mM).
Large single crystals were obtained through a fine crystallization optimization, varying PEG 8000 (6-11%) and bicine buffer pH. Crystals with different morphologies appeared in 2-5 days and crystals grew to full size within two weeks (Figure 6). Prior to X-ray diffraction data collection, crystals were soaked in cryoprotectant solution (reservoir buffer containing up 30% dimethyl sulfoxide). The optimized process resulted in quality crystals suitable for X-ray diffraction measurements at near atomic resolution.
X-ray diffraction data analysis
A crystal structure of the wild type α2β2 StTS complex was prepared with methods described in this article and X-ray diffraction data was collected at near atomic resolution. The crystal was soaked in cryoprotective solution containing F9 inhibitor (2- ({[4- (Trifluoromethoxy)Phenyl]Sulfonyl}Amino)Ethyl Dihydrogen Phosphate) and L-Tryptophan.
A complete X-ray diffraction data set was collected on the SIBYLS synchrotron beamline 12.3.1 at the Advanced Light Source (Berkeley-CA) by rotating the crystal 360° in increments of 0.5°. X-ray diffraction intensities were processed, and data-collection statistics are summarized in Table 1. Symmetry analysis indicates that the crystal belonged to the monoclinic space group C2. The unit-cell parameters are a = 182.55, b = 59.30, c = 67.37Å, α = 90.00, β = 94.82, γ = 90.00°. The calculated value of the Matthews coefficient (Vm = 2.57 Å3 Da-1) suggests the presence of one TS heterodimer molecule (αβ StTS) in the asymmetric unit of the crystal with a solvent content of 52.08%35,36. All X-ray data were collected at low temperatures (100 K) to improve the diffraction quality and decrease the radiation decay. The α2β2 StTS crystal structure in complex was solved by the molecular replacement method using the wild type αβ StTS model in complex with the inhibitor F9 at the α-site and cesium ion at the metal coordination site (PDB ID code: 4HT3). The final coordinate file and the structure factors were deposited in the PDB with accession code 5CGQ (Figure 7A). The crystal structure of the wild type α2β2 StTS complex with inhibitor F9 at the enzyme α-site (Figure 7B), cesium ion at the metal coordination site (Figure 7C), the cofactor pyridoxal 5'-phosphate covalently bonded to βLys87 (Figure 7D), and the product L-tryptophan at the enzyme β-site (Figure 7E) was solved at 1.18 Angstrom resolution. Model 5CGQ is the highest resolution α2β2 StTS crystal structure deposited in the PDB to date.
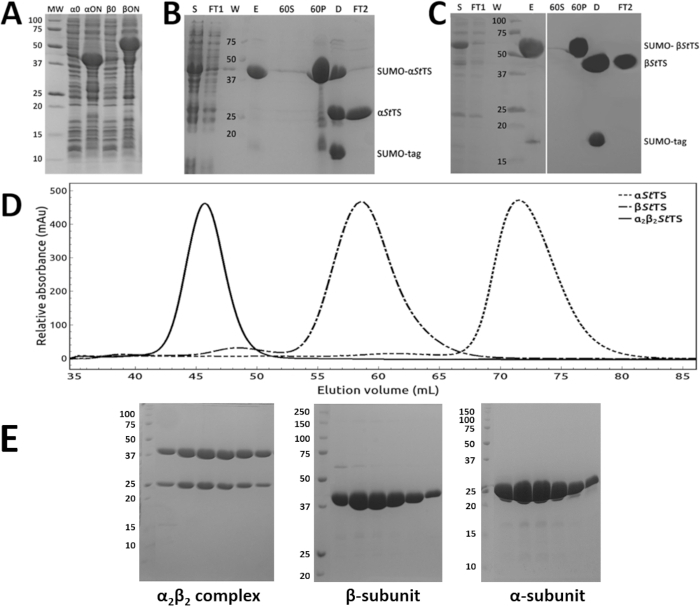
Figure 1: Purification of the α- and β-subunits and the α2β2StTS complex. (A) Recombinant protein expression. 12% SDS-PAGE gel of the overnight expression profile of SUMO-αStTS (αON) and SUMO-βStTS (βON) after IPTG induction at 30 °C (α/β0 prior IPTG induction). (B, C) Ni-NTA affinity chromatography followed by ammonium sulfate precipitation (60% saturation), (S) clarified crude extract (FT1) Ni-NTA column pass through sample (W) column wash sample (E) eluate sample (60S) and (60P) supernatant and precipitate fractions after high-speed centrifugation, respectively (D) SUMO-protease digestion product (FT2) Ni-NTA column pass through sample containing the tag-less αStTS or βStTS subunit. (D) elution profile of αStTS subunit (28.67 kDa), βStTS subunit (42.86 kDa), and α2β2 StTS complex (143.06 kDa) with a HiPrep 16/60 Sephacryl S-200 HR size exclusion chromatography column. Each run was performed separately. (E) SDS-PAGE gels of the excluded peak fractions from each individual chromatography. While 15% SDS-PAGE gels were prepared to analyze α2β2 StTS complex and αStTS subunit, a 12% SDS-PAGE gel was prepared to analyze the βStTS subunit. Lane MW, molecular-weight markers in kDa. Please click here to view a larger version of this figure.
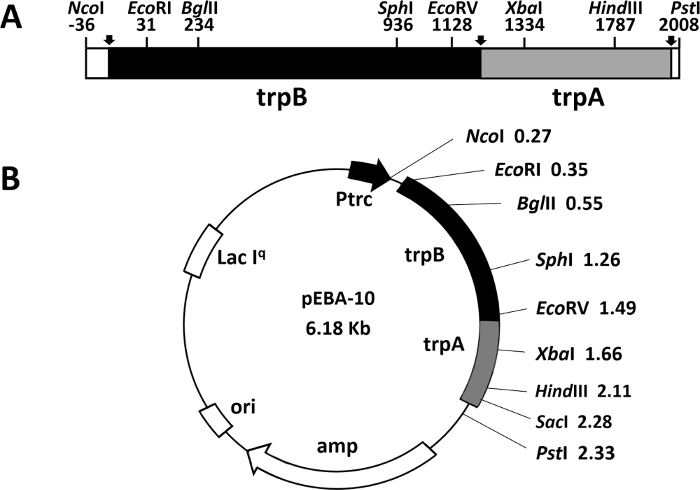
Figure 2: Representation of the construct pEBA-10. (A) Representation of the wild type translationally coupling gene (trpA and trpB) encoding the α– and β-subunits of the tryptophan synthase from bacterium Salmonella enterica serovar typhimurium (Yang, Ahmed et al. 1996). (B) The vector contains an ampicillin resistance (amp) gene, a replication origin (ori), a lacIq gene to better shutdown a lac promoter in absence of IPTG inducer, and the LacI-repressed promoter. Please click here to view a larger version of this figure.
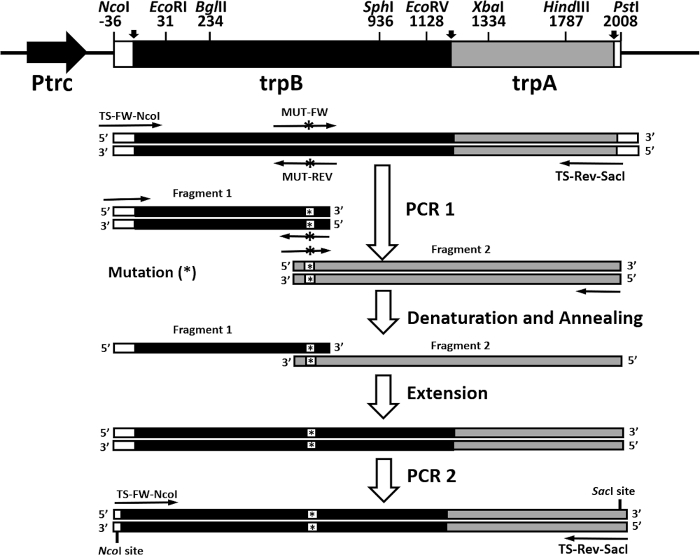
Figure 3: Overall representation of the two-step PCR mutagenesis protocol. The pEBA-10 vector was used as a DNA template. The first round of PCR was prepared with primers TS-FW-NcoI and MUT-REV (a reverse primer containing a mutation) to generate the first fragment and primers TS-Rev-SacI and MUT-FW (a forward primer containing a mutation) to generate the second fragment). Fragments were gel purified and equimolarly combined, heat denatured, and annealed. The recombinant strands were extended with polymerase and deoxyribonucleotides. The second round of PCR was prepared with primers TS-FW-NcoI and TS-Rev-SacI. Please click here to view a larger version of this figure.
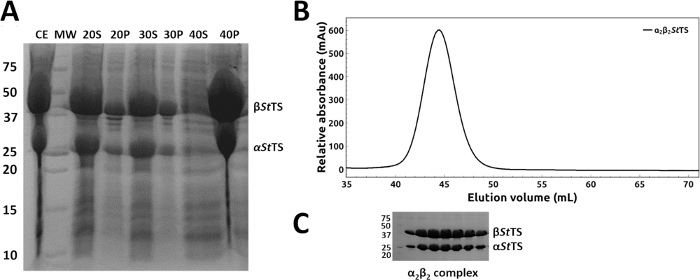
Figure 4: Purification of wild type and mutant form of α2β2 StTS complex. (A) 12% SDS-PAGE gel of samples collected along the ammonium sulfate precipitation using 20, 30, 40 and 50% ammonium sulfate saturation at room temperature: (CE) crude extract (S) and (P) supernatant and precipitate fractions after high-speed centrifugation. (B) Elution profile of α2β2 StTS (143.06 kDa) complex with a HiPrep 16/60 Sephacryl S-200 HR size exclusion chromatography column. (C) 12% SDS-PAGE gel picture of the excluded peak fractions. Lane MW, molecular-weight markers in kDa (Precision Plus Protein Unstained Standards, Bio-Rad). Please click here to view a larger version of this figure.
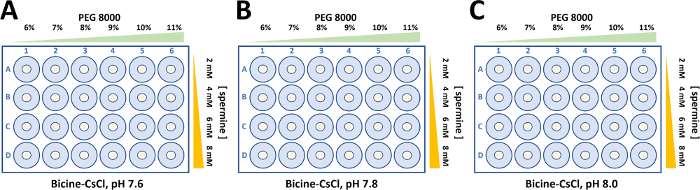
Figure 5: Crystallization optimization for wild type and mutant form of α2β2 StTS complex. Crystals were grown in 50 mM Bicine-CsOH buffer containing 50 mM CsCl2. The concentration of polyethylene glycol 8000 (6-11%) and spermine (2-8 mM) were varied to obtain single large crystals forms to perform structural studies by X-ray protein crystallography. (A) Bicine-CsOH, pH 7.6. (B) Bicine-CsOH, pH 7.8. (C) Bicine-CsOH, pH 8.0. Please click here to view a larger version of this figure.

Figure 6: Photomicrograph of crystals of wild type and mutant form of α2β2 StTS complex. Crystals differ in morphology, but they belong to the space group C2. The crystals grew to their full dimensions in the final conditions after two weeks. Crystals of approximately 0.20 x 0.15 x 0.10 mm in size. (A-D) PLP holo-crystals in complex with cesium ion at the metal coordination site of the wild type form (column A), mutant form α2β2 βQ114A (column B), α2β2 βK167T (column C), and α2β2 βS377A (column D). Please click here to view a larger version of this figure.
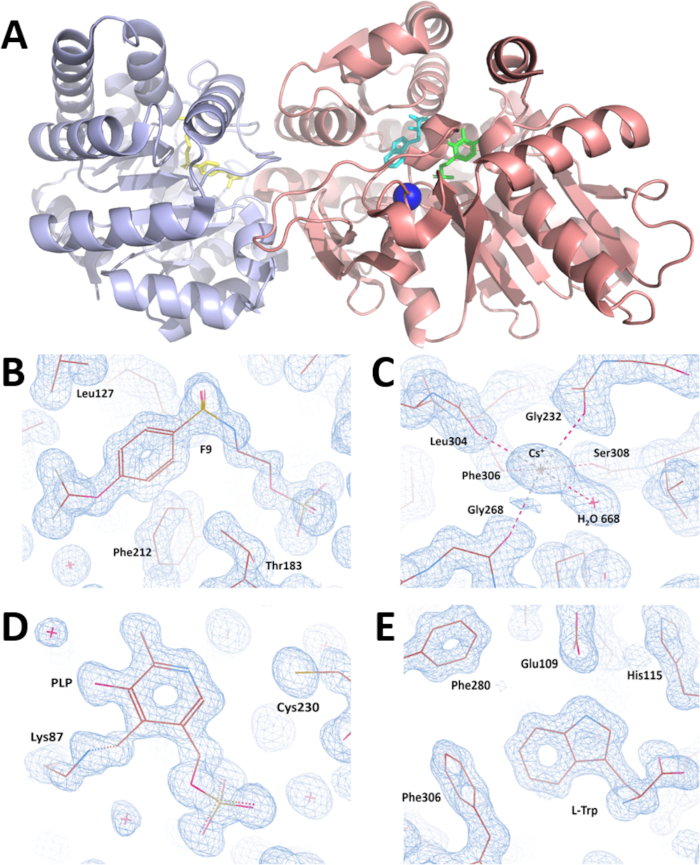
Figure 7: Overall visualization of crystal structure and validation of electron density maps obtained after crystal structure refinement. (A) crystal structure of the wild type α2β2 StTS complex with inhibitor F9 at the enzyme α-site (yellow colored), cesium ion at the metal coordination site (blue colored), the cofactor pyridoxal 5'-phosphate covalent bonded to βLys87 (green colored), and the product L-tryptophan at the enzyme β-site (cyan colored) at 1.18 Angstrom resolution. While the α-subunit is colored in light blue, the β-subunit is colored in salmon. (B-E) Electron density maps contoured at 1.0 r.m.s. level around (B) inhibitor F9 (C) cesium ion (D) pyridoxal-5′-phosphate, and (E) L-Tryptophan. Please click here to view a larger version of this figure.
| Data collection and processing | |
| X-ray source / Beam line | ALS Beamline 12.3.1 |
| Wavelength (Å) | 10,000 |
| Resolution (Å) | 40.00 – 1.18 (1.24 – 1.18) |
| Total number of reflections | 2151280 (252941) |
| Total number unique reflections | 231646 (32187) |
| Space group for indexing, scaling and merging | C 1 2 1 |
| Cell dimensions | |
| a, b, c (Å) | 182.55, 59.30, 67.37 |
| α, β, γ (°) | 90.00, 94.82, 90.00 |
| Mosaicity | 0.61 |
| Matthews volume VM (Å3 Da-1) | 2.57 |
| Rmeas (%) | 8.6 (93.0) |
| <I/σ(I)> | 14.7 (3.0) |
| CC1/2 (%) | 0.999 (0.778) |
| Completeness (%) | 98.6 (94.2) |
| Multiplicity | 9.3 (7.9) |
| Refinement statistics | |
| Rwork/Rfree (%) | 14.04 / 16.05 |
| RMSD bond length (Å) | 0.0120 |
| RMSD bond angle (°) | 14,059 |
| Ramachandran favored | 515 (96.44%) |
| Ramachandran allowed | 16 (3.00%) |
| Ramachandran disallowed | 3 (0.56%) |
Table 1: Data collection and processing. Values in parentheses are for the outer shell.
