The eukaryotic FAH domain-containing protein 1 (FAHD1) acts as bi-functional oxaloacetate (OAA) decarboxylase (ODx)1 and acylpyruvate hydrolase (ApH)2. It is localized in mitochondria2 and belongs to the broad FAH superfamily of enzymes1,2,3,4,5,6. While its ApH activity is only of minor relevance, the ODx activity of FAHD1 is involved in the regulation of the TCA cycle flux1,7,8,9. OAA is not only required for the central citrate synthase reaction in the tricarboxylic acid cycle but also acts as a competitive inhibitor of succinate dehydrogenase as part of the electron transport system and as a cataplerotic metabolite. Downregulation of FAHD1 gene expression in human umbilical vein endothelial cells (HUVEC) resulted in a significant reduction in the rate of cell proliferation10, and significant inhibition of mitochondrial membrane potential, associated with a concomitant switch to glycolysis. The working model refers to mitochondrial dysfunction associated senescence (MiDAS)11-like phenotype8, where mitochondrial OAA levels are tightly regulated by FAHD1 activity1,8,9.
Recombinant protein is easier to obtain via expression and purification from bacteria12 rather than from tissue. However, a protein expressed in bacteria may be biased by possible lack of post-translational modifications, or may simply be problematic (i.e., due to plasmid loss, bacterial stress responses, distorted/unformed disulfide bonds, none or poor secretion, protein aggregation, proteolytic cleavage, etc.). For certain applications, protein needs to be obtained from cell lysate or tissue, in order to include such modifications and/or to exclude possible artifacts. Purified protein from tissue supports the development of high-quality antibodies, and/or potent and specific pharmacological inhibitors for selected enzymes, such as for FAHD113.
This manuscript presents a series of methods for the extraction and purification of FAHD1 from swine kidney and mouse liver. The described methods require fast protein liquid chromatography (FPLC) but otherwise use common laboratory equipment. Alternative methods may be found elsewhere14,15,16,17. After total protein extraction, the proposed protocol involves a testing phase, in which sub-protocols for ammonium sulfate precipitation and ionic exchange chromatography are discussed (Figure 1). After defining these sub-protocols, the protein of interest is extracted via a sequential strategy using ionic exchange and size-exclusion chromatography with FPLC. Based on these guidelines, the final protocol may be adapted individually for other proteins of interest.
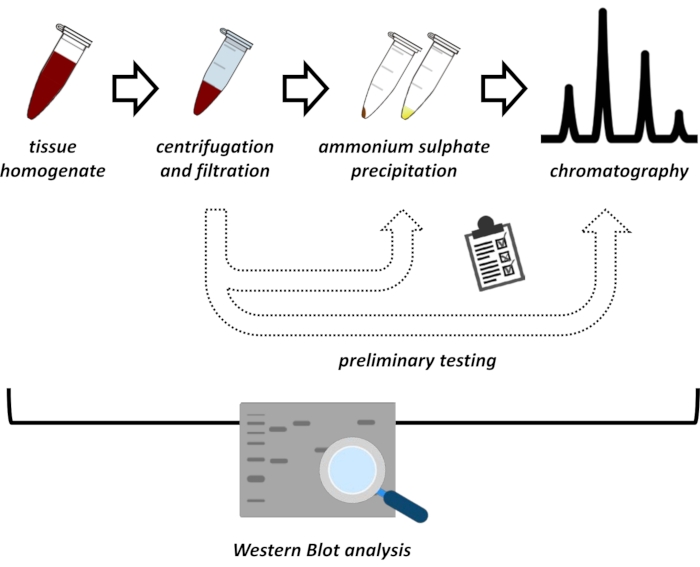
Figure 1: The overall strategy of this protocol. From top to bottom: Protein is extracted from tissues. Tissue homogenate is prepared, centrifuged, and filtrated. For each pair of supernatant and pellet-derived samples, tests for ammonium sulfate precipitation and ionic exchange chromatography (FPLC) need to be performed to probe for optimal conditions. After establishing these sub-protocols, the protein may be extracted via a sequential procedure of ammonium sulfate precipitation, ionic exchange chromatography, and repetitive size exclusion chromatography (FPLC) at varying pH and salt concentrations. All steps need to be controlled by western blot. Please click here to view a larger version of this figure.
FAHD1 protein was extracted from swine kidney and mouse liver using the presented protocol. For mouse tissue, multiple organs are required to obtain several µg after the final purification step. For this reason, this article focuses on the extraction of FAHD1 from swine kidneys, which is a much more exemplary experiment. The extraction of FAHD1 from the mouse liver is performed to present the difficulties and possible pitfalls of this protocol. It is generally recommended to use organs that show a high expression level of the protein one wants to purify. The Human Protein Atlas22 may be of help to estimate the expression in the model system, or one may perform preliminary western blot experiments with different crude tissue lysates to assess these levels. The positive control used in all western blot analyses was a tagged recombinant protein, running at a slightly higher molecular weight.
As a first experiment, the extraction of FAHD1 from the swine kidney is presented. The process of tissue homogenization (step 1) and total protein extraction (step 2) is presented in Supplementary Figure 1 (see the legend for a description of the individual steps). An aliquoted tenth of the total lysate was used for the following experiments.
Ammonium sulfate precipitation was tested once for this lysate, by adding ammonium sulfate to the lysate in different concentrations (step 4) (Figure 2). After centrifugation, pellets, and supernatants were sampled and analyzed with western blot using a defined polyclonal antibody2 raised against human FAHD1. 200 ng of recombinant His/S tagged human FAHD1 obtained from E. coli12 was loaded as the positive control. The FAHD1 protein band can be seen at 25 kDa, while the tagged positive control runs at 34 kDa. Based on this data, a protocol was defined where the lysate is treated with 25% ammonium sulfate, i.e., conditions at which FAHD1 is still in the supernatant, while the majority of other proteins is precipitated. This is a major step in the purification strategy. Ammonium sulfate precipitation is an effective cleaning step before proceeding with other methods. Of note, amounts of ammonium sulfate higher than 30% will not be used, because ammonium sulfate gradually distorts the quality of SDS-PAGE and western blot.
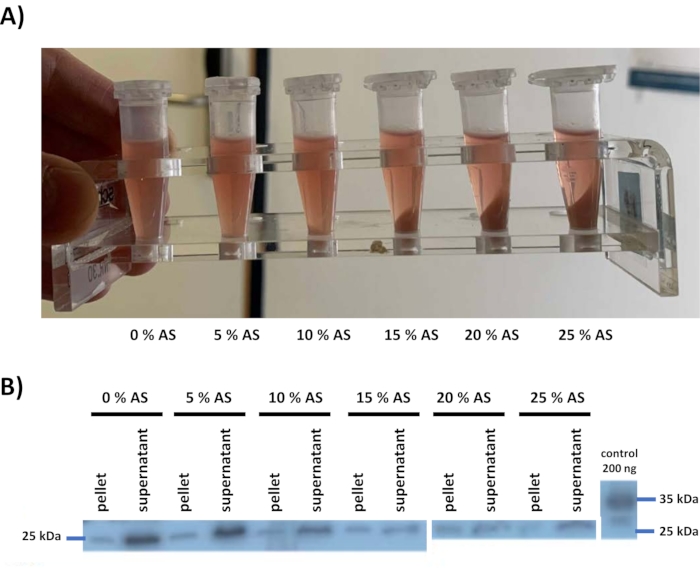
Figure 2: Ammonium sulfate precipitation and western blot screening for swine FAHD1. (A) Ammonium sulfate was added to the lysate in different concentrations (5% to 25% maximum) to precipitate proteins from the solution. (B) The presence of swine FAHD1 (25 kDa) was determined in the individual pairs of pellets and supernatant corresponding to varying ammonium sulfate concentrations via western blot using a polyclonal antibody raised against human FAHD1. The supernatant corresponding to 25% ammonium sulfate precipitation still shows the presence of swine FAHD1 in the supernatant, at the same time precipitating a good amount of other proteins. 200 ng of His/S-tagged recombinant protein was loaded as a positive control (34 kDa). Please click here to view a larger version of this figure.
After testing ionic exchange columns with FPLC, a strategy was defined where swine FAHD1 remains in the flow-through of an anionic exchange chromatography at pH 9 (step 5). Starting with lysate obtained by ammonium sulfate precipitation at 25% (as defined by previous testing experiments), anionic exchange chromatography was used to eliminate further proteins from the solution (Figure 3A). Binding proteins were removed by elution from the column, while the flow-through was further purified via SEC at pH 7.4 (Figure 3B). Following both steps, western blot analysis identified all the fractions that contained swine FAHD1, that were pooled and concentrated to 2 mL.
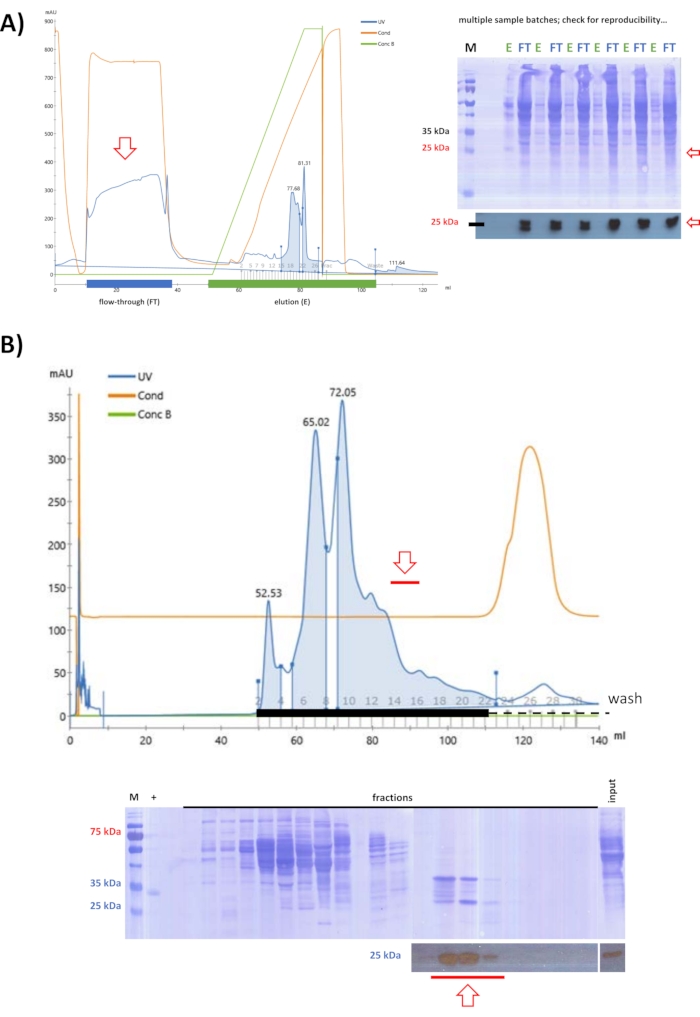
Figure 3: Purification of swine FAHD1 with FPLC – part 1. Red arrows in the chromatogram and blots indicate the presence of FAHD1. In the chromatogram, "UV" denotes the UV absorption at 280 nm, "Cond" denotes the conductivity (related to the salt concentration in the buffer), and "Conc B" denotes the percentage of high salt buffer in the buffer gradient (0% pure low salt buffer; 100% pure high salt buffer). (A) Purification of swine FAHD1 from lysate obtained by ammonium sulfate precipitation at 25% with an anionic exchange column (FPLC). While many proteins bind to the column, swine FAHD1 is found in the flow-through. Dirt and non-protein contaminants are removed by elution from the column, while the flow-through is further processed. (B) Flow-through from the previous anionic exchange chromatography in (A) is further purified via size exclusion chromatography (FPLC) at pH 7.4. (A,B) Western blot analysis (cropped blots at the bottom) identified fractions containing swine FAHD1, that are pooled and concentrated. The full blots depict the Coomassie-stained membrane after western blot analysis. Please click here to view a larger version of this figure.
The filtered sample was applied again to SEC but at buffer conditions at pH 9 (see Table 1) (Figure 4). The rationale behind this approach is that the pH and salt content of the mobile phase may influence the elution profile of globular proteins20, and an enhanced elution profile for FAHD1 was found under these buffer conditions. One fraction contained protein of adequate purity levels for basic enzyme activity assays12 to finally confirm the protein's identity.
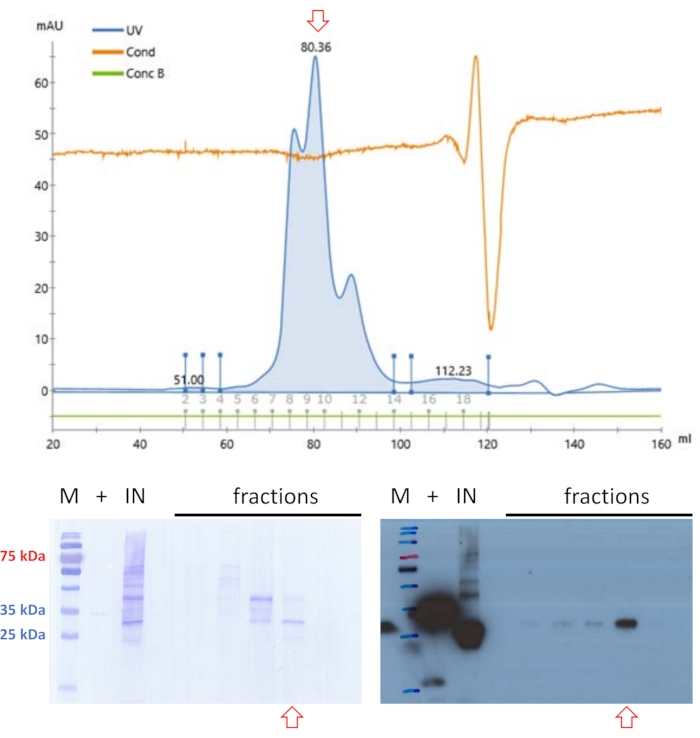
Figure 4: Purification of swine FAHD1 with FPLC – part 2. Red arrows in the chromatogram and blots indicate the presence of FAHD1. Protein samples previously purified with anionic exchange and size-exclusion chromatography are further purified via size exclusion chromatography (FPLC) at pH 9 (top panel). Western blot analysis depicted in the lower right identifies fractions that contain swine FAHD1, that are pooled and concentrated (bottom panels). The blot in the lower left shows the Coomassie staining of the membrane after the western blot. Please click here to view a larger version of this figure.
An alternate strategy (i.e., SEC followed by ionic exchange chromatography) was tested with 2 mL of the lysate obtained after ammonium sulfate precipitation (step 4) (Supplementary Figure 2). The rationale for this experiment is that by first using SEC, the FAHD1 containing fractions may be separated from the majority of other proteins, while a subsequent ionic exchange chromatography may be used to further purify the protein. Firstly, the lysate was fractionated using SEC at pH 7.4 (step 6). Western blot analysis identified all fractions that contained swine FAHD1. Secondly, these fractions were concentrated and further purified using an anionic exchange column at pH 9 (step 5). The chromatograms and western blot analysis (Supplementary Figure 2) show that this strategy is encouraging because the ionic exchange chromatogram shows a defined narrow peak that is associated with enriched protein levels in the western blot. However, the drawback of this strategy is that it is limited by the initial volume to be applied on the SEC (2 mL), so the throughput of this method is very low. Processing a whole swine kidney, for example, is not practical.
As another modification to this protocol, a cationic exchange column was included before the anionic exchange column when the swine FAHD1 was also present in the flow-through. At each step of the protocol, samples of the swine FAHD1 containing fractions were sampled and tested with an ODx assay, as described elsewhere12 (Supplementary Figure 3). The specific enzymatic activity increases along with the level of purity, i.e., along with the increasing relative amount of FAHD1 per total protein.
As a second example, the extraction of FAHD1 from mouse liver, i.e., a collection of liver tissue obtained from 20 mice is presented. Compared to the extraction of FAHD1 from the swine kidney presented above, this extraction proved to be more tedious. Problems were encountered at several steps of the protocol, which will be presented in the following. Total protein was extracted as described in step 2 (Supplementary Figure 4). As mouse liver homogenate tends to be a very sticky and slimy entity, filter papers (similar to coffee filter units) were used to pre-filter the lysate, which was diluted 1:3 in lysis buffer after extraction, to make the solution more like a liquid. This dilution enhanced the filtration procedure; however, it made the subsequent detection of the protein with western blot more tedious. After filtration, the prepared lysate had to be concentrated before further use. An aliquoted tenth of the total lysate was used for the following experiments.
A first testing step for the ammonium sulfate precipitation (Supplementary Figure 4I) displayed a possible problem that may be encountered. Ammonium sulfate at higher concentrations disrupts SDS-PAGE gels and causes a smile effect when run at 150 V (Supplementary Figure 5A; negative result). The same SDS-PAGE run at 80 V displays a positive result (Supplementary Figure 5B). In the latter case, the smile effect forms initially but is ultimately resolved because of the lower voltage applied. As the initial solution had to be heavily diluted to be able to filtrate the lysate, initial western blot analysis of these samples was unsuccessful, i.e., the positive control was detected by the antibody, but not the protein in the applied samples. This problem was solved using a higher concentration of both the lysate (concentrated using centrifugal filter units) and the antibody, and by applying the antibody overnight in the cold room while shaking. Ultimately, the western blot analysis provided both the pieces of information that the protein was present in the lysate, and that the protein was still present in the supernatant at 15% ammonium sulfate (Supplementary Figure 5C). Of note, SDS samples containing higher amounts of ammonium sulfate (>15%) precipitated while heating and the protein was lost. This can also be seen in the Coomassie staining and the western blot (Supplementary Figure 5C). This effect was not observed for the swine kidney, so it may be tissue-specific.
Following ammonium sulfate precipitation at 15%, 10 mL of lysate was applied to cationic exchange chromatography at pH 6.8. Western blot analysis displayed the mouse FAHD1 protein to be in the flow-through (Figure 5A). The eluted protein fraction seems to be minor; however, this example displays a secondary effect of ionic exchange chromatography. At this step of the protocol, the solution may still contain many compounds that might not be protein. Ionic exchange chromatography is a fast and easy way to get rid of such contaminations. Given that performing ionic exchange chromatography takes a few hours to perform (including all washing steps, the experiment itself takes half an hour), it provides a simple method of clearing the sample from non-protein contaminations.
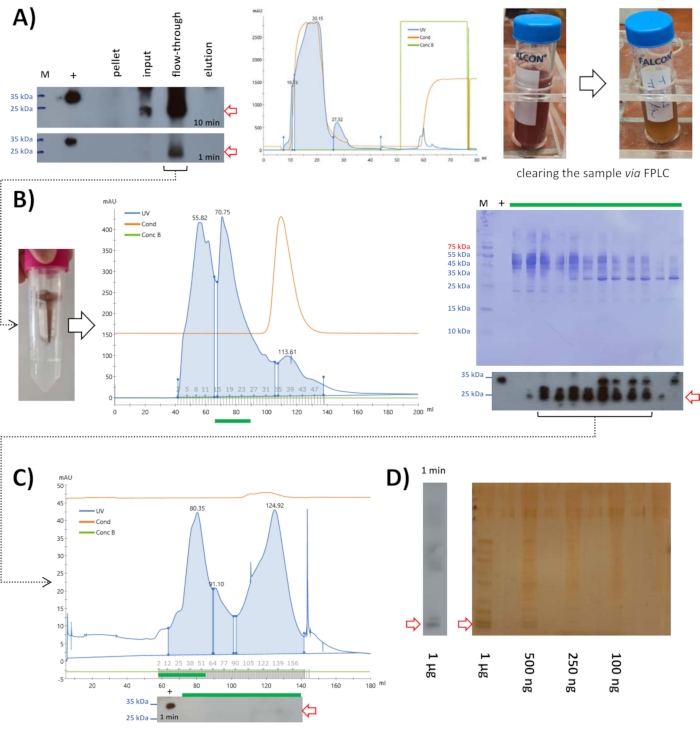
Figure 5: Purification of mouse FAHD1 with FPLC. Red arrows in the chromatogram and blots indicate the presence of FAHD1. (A) After ammonium sulfate precipitation at 15%, the sample was centrifuged, filtered (0.22 µm), and applied to a cationic exchange column. Western blot analysis shows that the protein is in the flow-through and the chromatogram shows that not much of the other proteins has been removed by this step. However, comparing the appearance and viscosity of the input with the flow-through shows that the collected flow-through is much clearer (right panel comparing input and flow-through). (B) The flow-through collected from the cationic exchange column was reduced to 2 mL via centrifugation filters and applied to size exclusion chromatography at pH 7.4. (C) The fractions containing FAHD1 were pooled, concentrated, and applied to a second round of size exclusion chromatography at pH 9. (D) The fractions containing FAHD1 were again pooled and applied to SDS-PAGE with silver staining, to see the remaining contaminations. Please click here to view a larger version of this figure.
The sample was further applied to size exclusion chromatography at pH 7.4 (Figure 5B). Western blot analysis displayed fractions containing the mouse FAHD1 protein, that were pooled and concentrated to 2 mL. This 2 mL concentrate was frozen at -20 °C with 10 µL of β-mercaptoethanol and thawed the next day. A precipitate was formed that was filtered via syringe filter units (0.22 µm). Samples for SDS-PAGE and western blot analysis were taken to assure that the mouse FAHD1 protein was still in solution.
Samples containing the protein (from western blot analysis) were pooled, concentrated via centrifugation filters, and applied to SDS-PAGE with silver staining to see the remaining contaminations (Figure 5D). This revealed that the protein solution is not highly pure and that the amounts of protein obtained via this method from mouse liver were not very high either (a few µg in total as determined using BCA assay kit; see Table of Materials). The protein yield was much higher in the case of swine FAHD1 (about 1 mg in this step). The level of purityof FAHD1 protein extracted from the swine kidney is about 80% (estimated after silver staining; data not shown). This purity may be further increased via, for example, affinity chromatography using a defined antibody. Such and other limitations of this protocol will be discussed in detail further below.
| Name of Buffer/Solution/Material | Composition | ||
| Coomassie destaining solution | 30% (v/v) EtOH; 5 % (v/v) HOAc; in ddH2O | ||
| Coomassie staining solution | 0.05% Coomassie Brilliant Blue R-250; 50 % (v/v) MeOH; 10 % (v/v) HOAc; in ddH2O | ||
| Mono Q high salt buffer | 20 mM Tris-HCl; 1 M NaCl; 10 % glycerol; in ddH2O; adjust pH to 9.0 with NaOH | ||
| Mono Q low salt buffer | 20 mM Tris-HCl; 15 mM NaCl; in ddH2O; adjust pH to 9.0 with NaOH | ||
| Mono S high salt buffer | 44 mM NaH2PO4; 6 mM Na2HPO4; 1 M NaCl; in ddH2O; adjust pH to 6.8 with HCl | ||
| Mono S low salt buffer | 44 mM NaH2PO4; 6 mM Na2HPO4; 15 mM NaCl; in ddH2O; adjust pH to 6.8 with HCl | ||
| Protein lysis buffer | 1x PBS (250 mL); 50 mM NaF; 1mM PMSF; 2 µg/mL aprotinin, 1 mM activated orthovanadate; in ddH2O | ||
| Rabbit Anti-hFAHD1 polyclonal antibody (affinity isolated) | Custom made; polyclonal anti-hFAHD1 antibody purified from rabbit serum and purified with FPLC according to reference 12 | ||
| Recombinant hFAHD1 protein western blot control | Western blot control protein expressed in E.coli and purified with FPLC according to reference 12. | ||
| SDS-PAGE 5x sample buffer | 300 mM Tris HCl; 500 mM DDT; 10% (w/v) SDS; 50% (v/v) glycerin; 0.05% (w/v) bromophenol blue; in ddH2O; adjust pH to 6.8 with HCl | ||
| SDS-PAGE resolving gel (12.5 %) for discontinuous PAGE | ddH2O (9.5 mL); 3 M Tris HCl (2.2 mL); 5.5 mL of 40% Acrylamide/Bis Solution (29:1 ratio); 20% SDS (175 µL); TEMED (17 µL); 10% ammonium persulfate (175 µL); cast before the stacking gel and let it polymerize; cast the stacking gel on top | ||
| SDS-PAGE running buffer | 25 mM Tris HCl; 190 mM glycine; 0.5 % (w/v) SDS; in ddH2O; adjust pH to 8.3 with NaOH | ||
| SDS-PAGE stacking gel (12.5 %) for discontinuous PAGE | ddH2O (3.8 mL); 1 M Tris HCl (630 µL); 500 µL of 40% Acrylamide/Bis Solution (29:1 ration); 20% SDS (25 µL); TEMED (5 µL); 10% ammonium persulfate (50 µL); cast after the resolving gel has polymerized; apply a gel comb to create wells | ||
| SEC (G75) running buffer | 15 mM Tris-HCl; 300 mM NaCl; in ddH2O; adjust pH to 7.4 with NaOH | ||
| Silver staining developing solution | 250 mM Na2CO3 in ddH2O; add 12 µL of 37% (v/v) formaldehyde to 100 mL before use | ||
| Silver staining fixing solution | 40% (v/v) EtOH; 10% (v/v) HOAc; in ddH2O | ||
| Silver staining incubation solution | 30% (v/v) EtOH; 500 mM NaOAc; 8 mM Na2S2O3; in ddH2O; optional: add 600 µL of 50% (v/v) glutaraldehyde per 100 mL before use | ||
| Silver staining silver solution | 0.1% (w/v) AgNO3; in ddH2O; add 25 µL of 37% (v/v) formaldehyde to 100 mL before use | ||
| Silver staining stop solution | 40 mM EDTA solution at pH 7.6; transfer 744 mg of EDTA into 40 mL ddH2O and then add NaOH to dissolve and adjust pH to 7.6. Finally, adjust the total volume to 50 mL. | ||
| Western blot blocking buffer | PBS with 0.1% (v/v) Tween 20 and 5% (w/v) skim milk powder; filtrated | ||
| Western blot transfer buffer (10x) | Tris-HCl 250 mM; glycine 1.92 M; in ddH2O; adjust pH to 8.3 with NaOH; store at room temperature | ||
| Western blot transfer buffer (1x) | Western blot transfer buffer (10x) 100 mL; 800 mL of ddH2O; 100 mL of MeOH; store at 4 °C | ||
| Western blot washing buffer (PBS-T) | PBS with 0.1% (v/v) Tween 20 | ||
Table 1.
Supplementary Figure 1: Total protein extraction from swine kidney. (A) Kidney tissue was cut using a scalpel on a prepared properly cleaned glass plate put on polystyrene foam to prevent sliding; (B) 50 mL tubes containing 20 mL of ice-cold lysis buffer with protein/protease inhibitors are incubated on ice; (C) kidney tissue pieces were put into the buffer until the volume reached about 30 mL; (D) kidney tissue was homogenized using ultrasound (i.e., sonication); (E) crude homogenate was centrifuged in a table-top centrifuge at medium speed for 30 min; (F) several samples were processed simultaneously; (G) after first centrifugation, the supernatant was transferred into centrifuge tubes (compare to E), and centrifuged at high speed (10,000 x g) for 30 min; (H) this centrifugation step yields another pellet and supernatant, which is transferred into 50 mL tubes; (I/J) the pre-lysate is sequentially filtered by 0.45 µm and 0.2 µm filter units, aliquoted into 10 mL batches, and snap-frozen with liquid nitrogen for subsequently storing at -80 °C. Western blot analysis is used to verify the presence of the protein in all samples (pellets, supernatants, filtered lysate). Please click here to download this File.
Supplementary Figure 2: Purification of swine FAHD1 with FPLC; alternate strategy. Red arrows in the chromatogram and blots indicate the presence of FAHD1. (A) Purification of swine FAHD1 from lysate obtained by ammonium sulfate precipitation at 25% with size exclusion chromatography (FPLC) at pH 7.4. Western blot analysis identified fractions that contain swine FAHD1, that were pooled and concentrated. (B) Protein samples previously purified with size-exclusion chromatography (panel A) were further purified via anionic exchange chromatography (FPLC). Western blot analysis identifies fractions that contain swine FAHD1, that were pooled and concentrated. Please click here to download this File.
Supplementary Figure 3: Specific enzyme activity of swine FAHD1. Measured specific enzyme activity as a function of increasing level of purity. The table compares relative enzyme activity (nmol/min) with specific enzyme activity (µmol/min/mg). The assay shows a constantly increasing activity of swine FAHD1 after every purification step. Compared to the recombinant His/S-human FAHD1 (green) obtained from E. coli.12, swine FAHD1 showed a slightly higher activity (red) (n = 3). Please click here to download this File.
Supplementary Figure 4: Total protein extraction from mouse liver. (A,B,C) Snap-frozen liver tissue from 20 mice and lysis buffer were prepared on ice in 50 mL tubes; (D,E,F) liver tissue was homogenized using ultrasound (i.e., sonication); crude homogenate was centrifuged in a table-top centrifuge at medium speed for 30 min; the supernatant was transferred into centrifuge tubes, and centrifuged at high speed (10,000 x g) for 30 min; (G,H,I) the pre-lysate is sequentially filtered by paper filters, 0.45 µm and 0.2 µm filter units, aliquoted into 10 mL batches, and snap-frozen with liquid nitrogen for subsequently storing at -80 °C; (J) ammonium sulfate was added to the lysate in different concentrations (5% to 30% maximum) to precipitate protein from the solution; (K) pellets obtained after ammonium sulfate precipitation (panel J) were resuspended in H2O to take samples for SDS-PAGE and western blot analysis. Please click here to download this File.
Supplementary Figure 5: Ammonium sulfate precipitation and western blot screening for mouse FAHD1. Red arrows in the blots indicate the presence of FAHD1. (A) SDS-PAGE with samples obtained after ammonium sulfate precipitation was run at 125 V. This gel displayed a drastic smile effect, and such a gel cannot be evaluated. This is an example of a negative result. (B) SDS-PAGE with the same samples (panel A) was run at 80 V until complete. There is no smile effect in this gel, and this is an example of a positive result. (C) SDS PAGE and western blot analysis of the samples obtained after ammonium sulfate precipitation. Samples at higher concentrations of ammonium sulfate may precipitate inside the sample buffer, as indicated. They are lost and cannot be detected anymore, either via Coomassie staining (left) or western blot analysis (right). The best concentration of ammonium sulfate, in this case, was determined to be 15%. Please click here to download this File.
