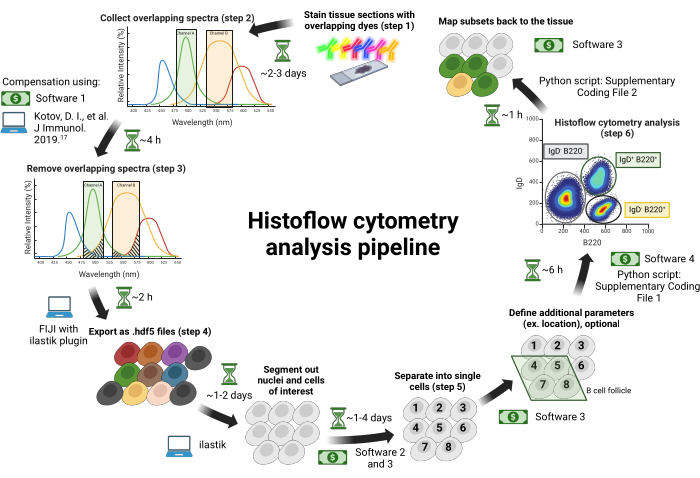
Figure 1: Histoflow cytometry workflow. Tissue sections are stained with spectrally overlapping dyes (step 1). Images are collected across individual excitation lasers paired with tunable bandpass filters to minimize spectral bleed-through between fluorophores (step 2). Spectral bleed-through between channels is corrected based on a compensation matrix generated using single color-stained tissue controls (step 3). Nuclei and cells in images are then identified using ilastik (step 4) and split into single cells in Software 3 (step 5). Additional parameters are incorporated before exporting all data from Software 3 into Software 4 to conduct flow cytometry analysis. Cell identities are then exported from Software 4 and mapped back to the cell IDs in the original tissue to resolve the spatial localization of cells in tissue sections (step 6). Money signs represent commercial software that requires purchase, and computer signs represent free software. The approximate time for each step is indicated beside the hourglass. Commercial software is listed in the Table of Materials. This figure is adapted from Jain et al.18. Please click here to view a larger version of this figure.
In Figure 3, lymph node tissues were stained with a rat anti-mouse CD138 antibody paired with a donkey anti-rat Dylight 405 secondary antibody, Nuclear Yellow, a rat anti-mouse CD4 antibody conjugated to Alexa Fluor 488, a rat anti-mouse IgD antibody conjugated to PerCP-eF710, a rabbit anti-mouse Iba1 antibody paired with a Donkey anti-rabbit Alexa Fluor 546 secondary antibody, a rat anti-mouse CD3ε antibody conjugated to PE-eF610, a rat anti-mouse B220 antibody conjugated with Alexa Fluor 647, and a rat anti-mouse CD45 antibody conjugated to APC-R700. A violet 405 nm laser was used to stimulate the Dylight 405 and Nuclear yellow dyes, and their emissions were detected in the ranges of 408-438 nm and 460-530 nm, respectively. A green 488 nm laser was used to stimulate the Alexa Fluor 488 and PerCP-eF710 dyes, and their emissions were detected in the ranges of 495-545 nm and 682-750 nm, respectively. A yellow-green 552 nm laser was used to stimulate the Alexa Fluor 546 and PE-eF610 dyes, and their emissions were detected in the ranges of 557-600 nm and 615-652 nm, respectively. A red 640 nm laser was used to stimulate the Alexa Fluor 647 and APC-R700 dyes, and their emissions were detected in the 647-682 nm and 700-750 nm ranges, respectively.
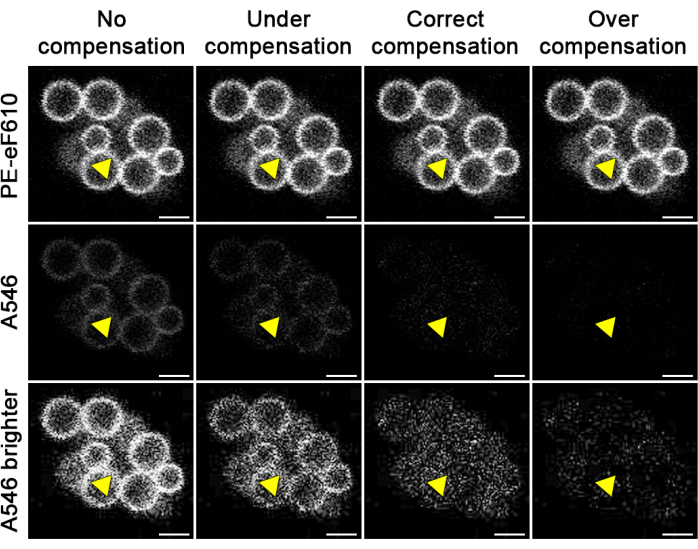
Figure 2: Examples of poor and quality compensation. Beads were stained with an antibody conjugated to PE-eFluor 610 and imaged in the channels corresponding to the PE-eFluor 610 (PE-eF610) and Alexa Fluor 546 (A546) channels. Different degrees of compensation were applied to the same image in the A546 channel (10% reduction for undercompensation, 28% reduction for correct compensation, and 60% reduction for overcompensation) to remove the bleed-through from the PE-eF610 fluorophore, resulting in different degrees of separation. To better visualize the degrees of compensation, a brightened version of the bleed-through images is provided. The yellow arrow points to the same area in each image corresponding to an area where there is real staining in the PE-eF610 channel. Scale bars: 4 µm. Please click here to view a larger version of this figure.
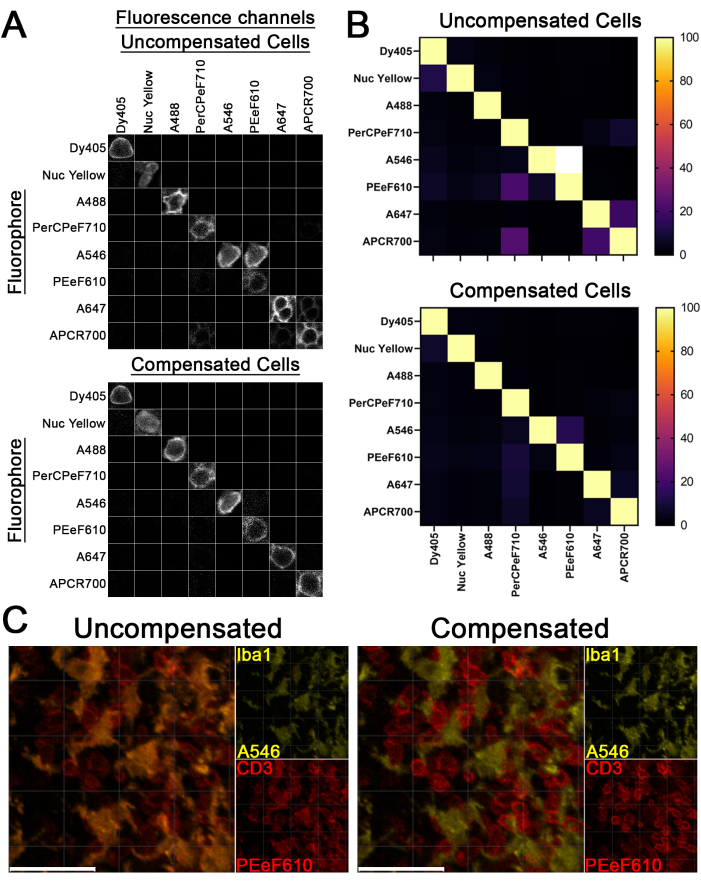
Figure 3: Fluorescence compensation using stained tissue sections. (A–C) Lymph node tissue sections were stained with single Abs conjugated to the listed fluorophores. Uncompensated single-stained samples were then imaged across all channels (A, top), and compensation was applied to the single-color controls (A, bottom). (B) Bleed-through between channels in (A) was quantified (mean fluorescence intensity of the current image divided by the mean fluorescence intensity of the channel of the current fluorophore) and presented in heatmaps. White squares represent values exceeding the 100% bleed-through. (C) Spectral overlap is assessed in the uncompensated and cell-based compensation conditions by comparing the spectral separation of Iba1-A546 and CD3-PEeF610 in fully stained samples. Scale bars: 30 µm. This figure is adapted from Jain et al.18. Please click here to view a larger version of this figure.
By following this protocol (summarized in Figure 1), spectrally overlapping fluorescent dyes should be effectively separated, as shown in Figure 3A,B. While complete separation of the dyes may not be possible, the separation should be sufficient to clearly differentiate different cell types (Figure 3C). If there are still shadows of spectrally overlapping cells in another channel, this may indicate that there is still bleed-through occurring, and the images may need reprocessing (Figure 2). Additionally, overcompensation will be apparent if the compensated area falls below the levels of fluorescence associated with background staining and will appear as black areas where the bleed-through once was (Figure 2). Overcompensation can lead to the loss of real signal in the channel to which compensation has been applied and must be avoided. Nonetheless, if this protocol has been followed correctly, there should be no issues with compensation, and thus, the researcher should consider whether any apparent bleed-through is artificial or a part of the biology.
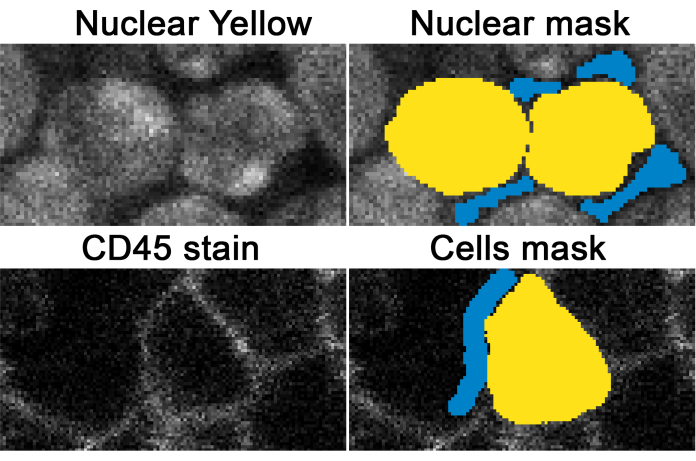
Figure 4: Example of an ilastik-based nucleus and cell detection. The nuclear yellow staining (top-left) was used to identify nuclei (top-right in yellow) and what is not a nucleus (in blue). Representative CD45 staining (shown in bottom-left) is used to identify cells (bottom-right in yellow) and what is not a cell (in blue). This figure is adapted from Jain et al.18. Please click here to view a larger version of this figure.
Once dyes are accurately separated, splitting the tissue into individual cells is essential for conducting single-cell analyses. The protocol described here uses user-defined machine learning (Figure 4) to separate cells even when they are closely compacted together, as shown in the lymph nodes shown in Figure 6.
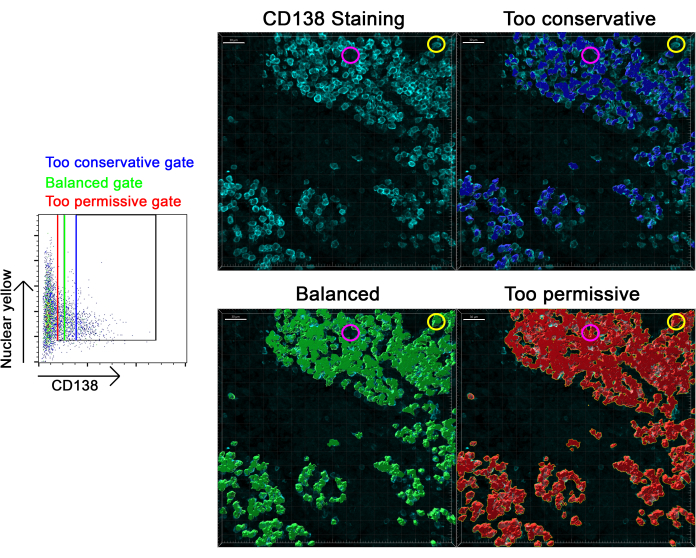
Figure 5: Evaluating whether a gate has been adjusted correctly. Representative CD138 staining (colored in cyan) in a lymph node is shown, and three separate gates (the dot plot to the left) were made to identify CD138+ cells in the tissue. An overly conservative estimate of CD138+ cells is shown as the blue gate and mapped to the tissue in blue. A balanced estimate of CD138+ cells is shown as the green gate and mapped to the tissue in green. An overly permissive estimate of CD138+ cells is shown as the red gate and mapped to the tissue in red. The yellow circle identifies an area where CD138+ cells are not being identified by the overly conservative gate, and the magenta circle identifies an area with no CD138+ cells, which is claimed to contain CD138+ cells by the overly permissive gate. Scale bars: 30 µm. Please click here to view a larger version of this figure.
Care should be taken when determining the precise positions of gates in the flow cytometry analysis (shown in Figure 5) to ensure that these gates are not overestimating or underestimating the abundance of cells. This should be determined by mapping the cells back onto the tissue, as in Figure 5, to see if the cells that do express your markers of interest are being captured in the analysis. If too many cells are being selected (for example, cells that do not express CD138 are being selected as being CD138+), then the gates should be shifted to be less permissive, and if too few cells are being selected, then the gates should be made more permissive. Gates should be optimized for selectivity such that the upstream gates are optimized first, then each subsequent gate in the chain is optimized next. In the end, effective optimization of a gating strategy should be capable of resolving cell subsets of interest (Figure 6 and Figure 7).
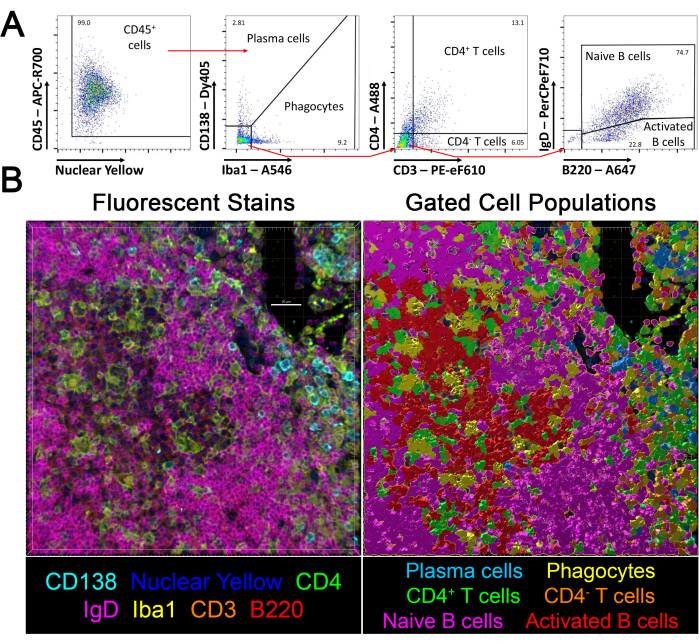
Figure 6: Flow cytometry-based identification of cells in tissue sections. (A) Fluorescent values exported from single cells from Software 3 in lymph node sections were analyzed in Software 4 to identify the stated cell populations based on the shown gating strategy. (B) Populations identified in Software 4 were mapped back onto the same tissue section, and surfaces were generated for each population. The CD45 stain is not shown. Scale bar: 30 µm. This figure is adapted from Jain et al.18. Please click here to view a larger version of this figure.
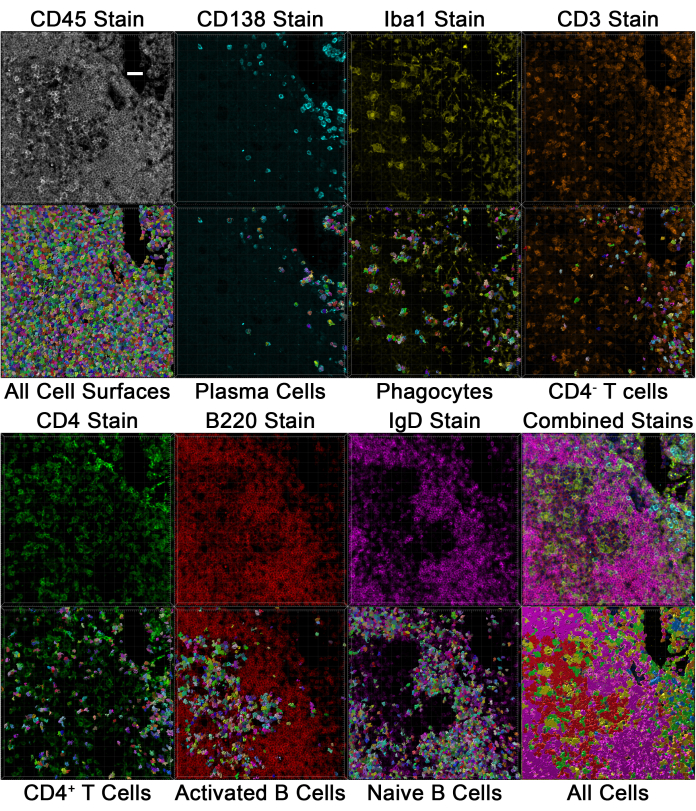
Figure 7: Breakdown of individual stains and populations in mouse lymph nodes. Individual stains for immune cell markers are shown in the top panels, and their associated gated population identified in Figure 6A is shown below each single stain. Scale bar: 30 µm, shown in the top left panel. This figure is adapted from Jain et al.18. Please click here to view a larger version of this figure.
This technique has been previously18 used to characterize immune cell infiltrates into spinal cords during neuroinflammation and validated that equivalent proportions of infiltrating immune cells are found using histoflow cytometry and flow cytometry. The technique has also been validated, demonstrating that it works in mouse spleens and brains and can identify differences between treatment conditions, which were reproduced in identical flow cytometry experiments (unpublished). This technique has also been used to analyze the spinal cords of mice receiving stereotactic injections of the CNS models.
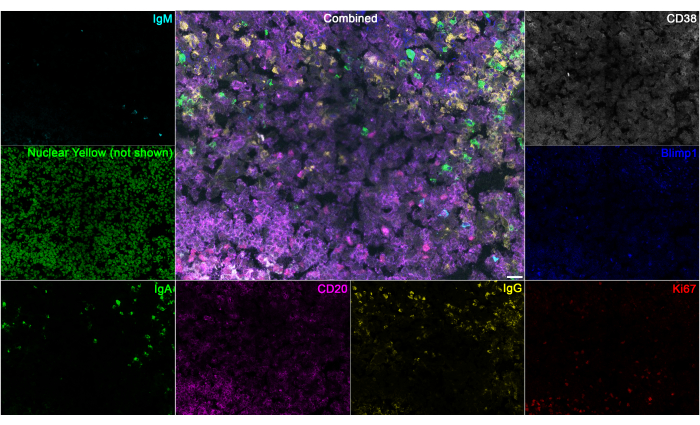
Figure 8: Separation of 8 dyes in a human tonsil section. Human tonsil sections were stained with CD20 to identify B cells and IgM, IgA, and IgG to identify plasma cells. Blimp1 and CD38 stains were incorporated to further validate plasma cell identities, and Ki67 to identify proliferating cells. The Nuclear yellow stain is not shown in the combined image. Scale bar: 22 µm. Please click here to view a larger version of this figure.
This technique can also be applied to analyze human tissue sections. As an example of staining within human tissues, human tonsil sections were stained with a selection of 8 markers: a goat anti-human IgM antibody conjugated to Dylight 405, Nuclear Yellow, a goat anti-human IgA antibody conjugated to Alexa Fluor 488, a mouse anti-human CD20 antibody paired with a goat anti-mouse F(ab')2 fragment conjugated to PerCP-eFluor 710, a goat anti-human IgG conjugated to Cy3, a rabbit anti-human Ki67 antibody paired with a goat anti-rabbit antibody conjugated to PE-Alexa Fluor 610, a rat anti-human Blimp1 antibody paired with an donkey anti-rat antibody conjugated to Alexa Fluor 647, and a mouse anti-human CD38 antibody conjugated to APC-R700. After compensation, the separation of these markers in the tonsil section can be seen (Figure 8). This demonstrates the utility of this technique in different tissues and species.
| Solution name | Composition | How to make | ||||
| Blocking buffer | 10% Horse Serum, 1% BSA, and 0.1% cold fish stain gelatin in PBS | Add 5 mL horse serum, 0.5 g BSA, 50 μL cold fish stain gelatin to 45 mL of PBS. Aliquot these solution into 15 mL tubes and store them at -20 °C until needed. | ||||
| Perm and blocking buffer | 10% Horse Serum, 1% BSA, 0.1% cold fish stain gelatin, 0.1% Triton X-100, 0.05% Tween-20 in PBS | Add 5 mL horse serum, 0.5 g BSA, 50 μL cold fish stain gelatin, 50 μL Triton X-100, and 25 μL Tween-20 to 45 mL of PBS. Aliquot these solution into 15 mL tubes and store them at -20 °C until needed. | ||||
| Perm and staining buffer | 1% BSA, 0.1% cold fish stain gelatin, 0.5% Triton X-100 in PBS | Add 0.5 g BSA, 50 μL cold fish stain gelatin, and 250 μL Triton X-100 to 50 mL of PBS. Aliquot these solution into 15 mL tubes and store them at -20 °C until needed. | ||||
| Quenching buffer | 5% Trueblack, 66.5% ethanol, 28.5% water | Preheat Trueblack solution at 70 °C for 5 min. Then add 50 μL of Trueblack to 950 μL of 70% ethanol to make the Trueblack buffer. Make this fresh each time.You will need ~500 μL per slide although the specific amounts will depend on the nature of your tissues. | ||||
| Staining buffer | 1% BSA and 0.1% cold fish stain gelatin in PBS | Add 0.5 g BSA and 50 μL cold fish stain gelatin to 50 mL of PBS. Aliquot these solution into 15 mL tubes and store them at -20 °C until needed. | ||||
Table 1: A list of the solutions and their compositions needed for staining tissue sections.
Supplementary Coding File 1: A Python script that collects the fluorescence values from each channel and creates one summary file. Please click here to download this File.
Supplementary Coding File 2: A Python script that creates a list of all the ID numbers of gated cells into a text file. Please click here to download this File.
