1. Solutions preparation
- Preparation of polydimethylsiloxane (PDMS) precursor solution
- Prepare the negative PDMS precursor solution (also used for the glue precursor solution). Scoop the elastomer into a weigh boat using a spatula and weigh it. Add the curing agent to the elastomer base at a 1:10 ratio. Mix the PDMS and curing agent gently and thoroughly using the spatula in the plastic weigh boat.
NOTE: This PDMS precursor solution is poured into the 6-well square pyramidal microwell plate to form the negative mold. This is the same solution that is used for the glue precursor solution. - Prepare the positive PDMS precursor solution. Scoop the elastomer base into the weigh boat using a spatula and weigh it. Add the curing agent to the elastomer base at a 1:9 ratio. Mix the PDMS and curing agent gently and thoroughly using the spatula in the plastic weigh boat.
NOTE: This PDMS precursor solution is later poured onto the negative mold to form the positive mold.
- Prepare the negative PDMS precursor solution (also used for the glue precursor solution). Scoop the elastomer into a weigh boat using a spatula and weigh it. Add the curing agent to the elastomer base at a 1:10 ratio. Mix the PDMS and curing agent gently and thoroughly using the spatula in the plastic weigh boat.
- Preparation of 0.3 M triethanolamine (TEA) buffer of pH 8
- Pipette 1 mL of TEA and 9 mL of 1x phosphate-buffered saline (PBS) to a 50 mL conical using a pipette aid to create a 0.75 M TEA solution. Titrate the solution to a pH of 8 using 1 N HCl or 1 N NaOH. Then, add enough 1x PBS to achieve a final volume of 25 mL to achieve a final TEA concentration of 0.3 M with a pH of 8.
CAUTION: Store HCl and NaOH solutions in a flammable cabinet at room temperature (RT). Wear personal protective equipment when handling.
- Pipette 1 mL of TEA and 9 mL of 1x phosphate-buffered saline (PBS) to a 50 mL conical using a pipette aid to create a 0.75 M TEA solution. Titrate the solution to a pH of 8 using 1 N HCl or 1 N NaOH. Then, add enough 1x PBS to achieve a final volume of 25 mL to achieve a final TEA concentration of 0.3 M with a pH of 8.
- Preparation of complete media
- To prepare the complete media, add 10% (w/v) or 56 mL of fetal bovine serum and 1% (w/v) or 5.6 mL penicillin and streptomycin to 500 mL RPMI medium.
- Place the solution at 37 °C for 10-20 min or until the solution is warm prior to use.
- Store the solution at 0-4 °C for up to 6 months.
- Preparation of 20% (w/v) polyethylene glycol (PEG) stock solutions
NOTE: Calculation is based on 100 µL solutions which could be scaled up or down as needed.- To prepare a 100 µL stock solution of 20% w/v 4-arm PEG-Acrylate (4-arm PEG-Ac), weigh 20 mg of 4-arm PEG-Ac powder in a microfuge tube. Add 70 µL of 0.3 M TEA buffer, then vortex the solution for about 30 s or until fully dissolved. Account for volume change due to powder dissolution by adding enough TEA buffer (~27 µL) to reach a final solution volume of 100 µL.
- To prepare a 100 µL stock solution of 20% w/v PEG-diSH, weigh 20 mg of PEG-diSH powder in a microfuge tube. Add 70 µL of TEA buffer, then vortex the solution for about 30 s or until fully dissolved. Account for volume change due to powder dissolution by adding enough TEA buffer (~27 µL) to reach a final solution volume of 100 µL.
NOTE: The PEG powder is very hygroscopic and needs to be stored in a desiccated container at -20 °C. When taking it out of the freezer, allow the PEG powder to thaw for 10 min before opening the bottle to weigh the powder. Purge the bottle with an inert gas such as nitrogen or argon to displace the moist air prior to returning it to the freezer. The stock solution of 4-arm PEG-Ac can be stored at 4 °C for up to 2 weeks prior to use. The stock solution of PEG-diSH needs to be prepared immediately prior to use and cannot be stored because the thiol groups react with each other to form disulfide bonds.
- Preparation of 2% v/v basement membrane matrix solution
- To prepare a 2% v/v basement membrane matrix working solution, add 20 µL of the basement membrane matrix (LDEV-free) to 9.98 mL of complete media and mix thoroughly by pipetting up and down ~10 times. Prepare the solution at 4 °C (on ice), then warm it to 37 °C (in an oven or incubator) and use immediately.
NOTE: The basement membrane matrix will start to form a gel at >10 °C, so be sure to mix the basement membrane matrix solution with complete media at 2-6 °C. The complete media composition is described in step 1.3.
- To prepare a 2% v/v basement membrane matrix working solution, add 20 µL of the basement membrane matrix (LDEV-free) to 9.98 mL of complete media and mix thoroughly by pipetting up and down ~10 times. Prepare the solution at 4 °C (on ice), then warm it to 37 °C (in an oven or incubator) and use immediately.
- Preparation of the cell fixative solution
- To prepare 1 mL of cell fixative solution containing 4% w/v of paraformaldehyde and 0.1% v/v of nonionic surfactant, first mix 891 µL of 1x PBS and 108 µL of paraformaldehyde (37% w/v concentration), and then add 1 µL of nonionic surfactant (100% concentration). Mix the solution thoroughly.
NOTE: Fixative solution should be made fresh every time fixation is performed.
CAUTION: Paraformaldehyde is flammable and may form combustible dust concentrations in the air. It causes skin irritation and serious eye damage. Avoid breathing in since it may cause respiratory irritation. Handle paraformaldehyde in a chemical fume hood and wear personal protective equipment. Wash hands thoroughly after handling. The nonionic surfactant causes skin irritation and serious eye damage. Wear protective gloves and eye protection or face protection when handling. To avoid release into the environment, open the bottle in a chemical fume hood. Wash hands thoroughly after handling.
- To prepare 1 mL of cell fixative solution containing 4% w/v of paraformaldehyde and 0.1% v/v of nonionic surfactant, first mix 891 µL of 1x PBS and 108 µL of paraformaldehyde (37% w/v concentration), and then add 1 µL of nonionic surfactant (100% concentration). Mix the solution thoroughly.
- Preparation of the staining solutions
- To prepare 3 mM of 3,3'-dihexyloxacarbocyanine iodide (DiOC), mix 2.65 mg of DiOC in 1 mL of DMSO.
- To prepare 1.5 mM of propidium iodide solution (PI), mix 1 mg of PI in 1 mL of de-ionized water.
- Preparation of the spheroid-clearing solution
- Prepare clearing solutions of 20%, 40%, and 80% v/v of formamide in 1x PBS for spheroid clearing.
- To make 10 mL of 20% v/v formamide, mix 8 mL of 1x PBS followed by 2 mL of formamide. To make 10 mL of 40% v/v formamide, mix 6 mL of 1x PBS followed by 4 mL of formamide. To make 10 mL of 80% v/v formamide, mix 2 mL of 1x PBS followed by 8 mL of formamide.
- After combining formamide and 1x PBS, mix the solution by vortexing for about 30 s.
- Prepare clearing solutions of 20%, 40%, and 80% v/v of formamide in 1x PBS for spheroid clearing.
2. Fabrication of square pyramidal microwells
- Fabricate negative PDMS mold of square pyramidal microwells as shown in Figure 1.
- Prepare 2 g (~1 mL) of negative PDMS precursor solution and pour it onto one well of a 6-well square pyramidal master mold. Note that 1 mL completely covers one well of the plate. After covering the master mold with PDMS, degas the PDMS precursor solution for 30 min by placing the 6-well square pyramidal plate in a vacuum desiccator. Then, cure the PDMS by placing the plate into a 60 °C oven for 24 h.
NOTE: Ensure the plate lid is removed for degassing and placed back on for curing. Degas the solution in a vacuum desiccator or through purging with an inert gas such as nitrogen or argon. If the negative mold precursor solution still has bubbles after 30 min, indicating inadequate degassing, place it in a vacuum desiccator for an additional 30 min. Use one or multiple plate wells simultaneously to prepare one or more PDMS negative molds. Square pyramidal microwells of different sizes can be used, such as 400 and 800 µm side lengths, as shown in Table 1. The same amount of PDMS is used regardless of the square pyramidal sizes. - Once PDMS cures while still warm, carefully remove the negative PDMS mold from the master mold using a spatula and cut the negative mold in a 35 mm in diameter slab using a biopsy punch. Place in a Petri dish and cover with the lid and allow for continued curing RT for an additional 24 h.
NOTE: To remove negative molds, use a spatula to get between the well plate and the PDMS mold and gently pull the negative mold from the master mold. The mold is cut into 35 mm slabs to fit a 35 mm Petri dish. Molds can be made in other sizes to fit plates of different diameters. The 35 mm negative mold slabs can be stored, protected from dust at RT, and reused for 6 months.
- Prepare 2 g (~1 mL) of negative PDMS precursor solution and pour it onto one well of a 6-well square pyramidal master mold. Note that 1 mL completely covers one well of the plate. After covering the master mold with PDMS, degas the PDMS precursor solution for 30 min by placing the 6-well square pyramidal plate in a vacuum desiccator. Then, cure the PDMS by placing the plate into a 60 °C oven for 24 h.
- Prepare positive PDMS mold of square pyramidal microwells.
- Place the 35 mm slabs of the negative PDMS mold into a 35 mm Petri dish with the textured microwells facing up.
- Prepare 2.5 g (~1.2 mL) of positive PDMS precursor solution as above and pour it onto the negative mold in the 35 mm Petri dish to completely cover the negative mold. Then degas the precursor solution for 30 min as above and place it into 60 °C oven for 3-4 h.
NOTE: Because PDMS is viscous, a bubble can form from air trapped under the negative mold. If a bubble forms under the negative mold, push the mold down gently using a spatula to release the bubble. If air bubbles remain, continue degassing for 30 min, or take a spatula and gently stir the positive mold solution until the bubbles pop. - Once the positive PDMS mold cures, remove the molds from the 35 mm Petri dish and immediately peel the positive mold from the negative mold.
NOTE: Timing is important for successful peeling of the positive mold. Removal is best done by slightly cutting into the positive mold using a razor to expose the interface between the positive and negative mold and peeling the molds from each other. Then peel away the edges of the circular mold. Gently peel the negative mold from the positive mold.
- Glue the molds to the bottom of the wells of a 48-well plate.
NOTE: Here, a 48-well plate is used, but other plates can be used as long as the mold slabs are cut into the correct diameters (for example, 6 mm in diameter for a 96-well plate).- Cut the positive molds into slabs using a 10 mm biopsy punch.
NOTE: Approximately 4 molds (each 10 mm in diameter) can be cut from one 35 mm diameter positive mold. - To glue the molds to the bottom of a 48-well plate, prepare the PDMS glue precursor solution (~0.5 mL or 1 g) as previously described27. Use tweezers to gently dip the flat side (not the side with the pattern of microwells) of the 10 mm positive mold into the PDMS precursor solution. Carefully place one mold per well of a 48-well plate, and gently press each mold to the well bottom using the tweezer. Place the assembled plate in a 60 °C oven for 4-24 h to allow the PDMS glue to cure.
NOTE: If PDMS glue precursor solution gets on the positive mold microwells, it can be wiped off using soft tissue paper, and the step can be repeated. When gluing, make sure that the glue does not cover the microwells. - Sterilize molds by adding 300 µL of 70% ethanol into each well of the 48-well plate using a 1000 µL pipette. Aspirate the 70% ethanol and place the 48-well plate uncovered in a tissue culture hood under UV (302 nm) for 2 h.
NOTE: Molds can be used for 6 months and re-sterilized as needed.
- Cut the positive molds into slabs using a 10 mm biopsy punch.
3. Multicellular tumor spheroid formation, harvest, and encapsulation in hydrogels
NOTE: The protocol outlined in this section is for U87 human glioblastoma cell line (see Figure 1 and Figure 2), but a similar protocol could be used with other cancer cell types.
- Multicellular tumor spheroid formation
- Wash microwell molds first with an anti-adherence rinsing solution by adding 300 µL of the solution to each well using a 1000 µL pipette. Then, centrifuge at 1620 x g for 3 min and aspirate the solution using a vacuum pump and a Pasteur pipette.
NOTE: Perform this step immediately prior to cell seeding. - Expose the cells to ~80 µL of 0.25% trypsin/EDTA per each cm2 of the culture flask area for 5 min at 37 °C. For example, 1 mL of trypsin/EDTA is appropriate for a T-25 cell culture flask. Neutralize the trypsin by adding the same volume of complete cell culture medium. For example, add 1 mL of complete medium to the trypsin-containing T-25 cell culture flask. Collect the cells from the tissue culture flask.
- Transfer 10 µL of the cell suspension into each port of a hemocytometer for cell counting. Use an inverted microscope to count the total number of cells and average that cell count from at least 8 quadrants, ensuring that the cell number in each hemocytometer quadrant is 20-50 for good cell count results. Multiply the calculated number by 104 to determine the final cell concentration.
- Resuspend the collected cells in the complete RPMI cell culture medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin at a desired final cell concentration, depending on the desired spheroid size, as shown in Table 1.
NOTE: The 800 µm microwells will yield ~75 spheroids in one well of a 48-well plate, and the 400 µm microwells will yield ~300 spheroids in one well of a 48-well plate. - Place 500 µL of cell suspension at the desired concentration in microwells and centrifuge the plate at 1620 x g for 3 min. Place the plate in a humidified incubator at 37 °C and 5% CO2 for 24 h to allow spheroids to form.
NOTE: If spheroids do not form, 2% v/v of the basement membrane matrix combined with complete media can be used to resuspend cells (more details in step 1.5).
- Wash microwell molds first with an anti-adherence rinsing solution by adding 300 µL of the solution to each well using a 1000 µL pipette. Then, centrifuge at 1620 x g for 3 min and aspirate the solution using a vacuum pump and a Pasteur pipette.
- Spheroids harvesting
- Using a 1000 µL pipette, firmly pipette 500 µL of complete medium into the well. Using 500 µL of medium from the well, flush the four quadrants of the well (specifically the top, bottom, left and right quadrants) by pipetting up and down at the quadrants three to four times to dislodge the spheroids. Gently aspirate the medium containing the spheroids (~1000 µL total) into a microcentrifuge tube using a 1000 µL pipette and allow the spheroids to settle to the bottom.
- Remove the supernatant and resuspend the spheroids to the desired final concentration. For example, to achieve ~8 spheroids in a 20 µL gel after encapsulation, resuspend the spheroids in 100 µL of media, yielding a spheroid concentration of ~75 spheroids/100 µL in the spheroid suspension.
- Spheroids encapsulation in hydrogels, as shown in Figure 2.
- To create 100 µL of a 10% w/v PEG hydrogel precursor solution, combine 50 µL of the spheroid suspension, followed by 30 µL of 20% w/v 4-arm PEG-Ac and finally 20 µL of 20% w/v PEG-diSH in a microcentrifuge tube. This will give a stochiometric molar ratio of acrylate (Ac) to thiol (SH) groups ensuring optimal crosslinking. Mix the gel precursor solution by pipetting up and down ~10 times.
NOTE: Hydrogel composition, volume, and polymer concertation can be changed as needed. The resultant hydrogels will be slowly degrading and non-cell adhesive. To make the hydrogel cell adhesive, an adhesive ligand such as RGDS can be added. To make the hydrogel enzymatically degradable, an enzymatically degradable peptide crosslinker containing cysteine residues on both ends could be added. When transferring spheroids, gently pipette the solution twice to dislodge spheroids and bring them into suspension to ensure even distribution of spheroids. - Pipette 20 µL of the gel precursor solution in between two parafilm-lined glass slides separated with 1 mm silicon spacers, and place slides with gel precursor solution in 37 °C, 5% CO2 incubator for 15 min to allow for gelation.
NOTE: Ensure the two glass slides are covered in parafilm to create a hydrophobic surface allowing for easy peeling upon gelation. Instead of parafilm, a hydrophobic coating solution can be used. A 20 µL volume of hydrogel precursor solution will result in a hydrogel slab of ~6 mm in diameter and 1 mm in height prior to swelling. Any spacer type and thickness can be used, but it is recommended that the gel thickness is kept at or below 1 mm (thicker hydrogels could limit oxygen diffusion and transport of nutrients to the cells) but larger than the spheroid diameter (so that spheroids are completely encapsulated in the gel). Any volume of the hydrogel precursor solution can be used. The 20-30 µL gels are suitable for a 24-well plate. - Once hydrogel gelation is complete, separate the two glass slides and gently peel the gels off the glass plate using a spatula. Place the gels into a 24-well plate, one per well, ensuring the surface containing the spheroids faces up.
NOTE: Spheroids will fall to the bottom of the gel during gelation, so inverting them for culturing will ensure that the spheroids are near the surface of the hydrogel for better access to nutrients and oxygen. Gelation can be monitored by observing any hydrogel precursor solution remaining in the microcentrifuge tube and not used to create slabs by inverting the tube and noting the time the gel stops flowing. - Add complete medium (~500 µL) to each well and ensure the hydrogel is submerged completely. Place the multiwell plate in a humidified incubator at 37 °C and 5% CO2 and culture the cells with medium changes every 2-3 days.
NOTE: Hydrogels can be cultured for up to 4 weeks or until the hydrogels degrade, changing the media every other day.
- To create 100 µL of a 10% w/v PEG hydrogel precursor solution, combine 50 µL of the spheroid suspension, followed by 30 µL of 20% w/v 4-arm PEG-Ac and finally 20 µL of 20% w/v PEG-diSH in a microcentrifuge tube. This will give a stochiometric molar ratio of acrylate (Ac) to thiol (SH) groups ensuring optimal crosslinking. Mix the gel precursor solution by pipetting up and down ~10 times.
4. Fluorescent staining
- Cell viability
- Use the stain 3,3'-dihexyloxacarbocyanine iodide (DiOC), which stains the mitochondria and endoplasmic reticulum of all cells, to determine cell viability. Use DiOC (3 mM) at a concentration of 0.02 µg/mL. Specifically, use a 20 µL pipette to add 2 µL of DiOC per every 1000 µL of media into the flask culturing the dissociated cells (at least 24 h prior to the formation process of spheroids in section 3). Allow 24 h for the DiOC to stain the cells.
- Use the nuclear and chromosome stain, propidium iodide, PI (1.50 mM), which enters only dead cells. To stain the cells, first aspirate all media and rinse the gel by using a 1000 µL pipette to add 500 µL of 1x PBS so the gel is completely submerged.
- Aspirate the PBS and add 500 µL of fresh media, followed by 30 µL of the PI solution to each well (i.e., 6 µL per every 100 µL of media). Cover the well plate with aluminum foil to protect it from light. Place the well plate in an incubator at 37 °C and 5% CO2 and allow 30 min for the PI to stain the dead cells.
- Remove the foil and aspirate the media from the wells. Use a 1000 µL pipette to add 500 µL of 1x PBS to submerge the hydrogel. Aspirate the 1x PBS and repeat the rinse two additional times. Add 500 µL of media to each well and image under a fluorescent inverted or confocal microscope.
- Calculate the cell viability by comparing the area of DiOC (all cells) to PI (dead cells), as represented in equation 1, using z-stack images from a confocal microscope or an inverted fluorescent microscope.
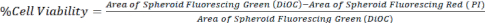 Eq. 1.
Eq. 1.
5. Immunofluorescence fixation, staining, clearing, and imaging of encapsulated spheroids
- Fixation and staining
- Aspirate the media from the wells where the hydrogels are cultured, and rinse the hydrogels by pipetting 500 µL of 1x PBS directly onto the hydrogels. Gently aspirate the 1x PBS.
- Fix the spheroids in the 24-well plate by using a 1000 µL pipette to add 500 µL volume of fixative solution per well. Allow the fixative to soak the gels for 30 min at RT. Remove the fixative solution using a 1000 µL pipette and discard in a designated waste container.
- Rinse the hydrogels by adding 500 µL of 1x PBS to each well. Aspirate the 1x PBS using a 1000 µL pipette and repeat the PBS rinse two additional times. Store the well plate in 500 µL of 1x PBS per well at 4 °C for up to 1 week or use immediately.
NOTE: Be careful not to pull the hydrogels into the pipette when aspirating PBS and fixative solution. Do so by tipping the plate to a ~45-degree angle, which will aid in seeing the hydrogels and preventing accidental aspiration.
CAUTION: Formaldehyde is toxic upon inhalation and contact. Handle with gloves in a chemical fume hood. - To stain the cells, incubate the hydrogel-encapsulated spheroids with primary antibodies for Nestin (200 µg/mL) and SOX2 (200 µg/mL) at a dilution of 1:200 of antibody: PBS. Use a 1000 µL pipette to aspirate the 1x PBS from the wells. Add 50 µL of the diluted antibody to each well. Allow 24 h for staining to be completed. Then, remove the staining solution using a 1000 µL pipette and discard the waste appropriately.
- Use a 1000 µL pipette to add 500 µL of 1xPBS, which is enough to submerge the hydrogel. Aspirate the PBS and repeat it two more times. Store the stained and submerged hydrogel in 1x PBS at 4 °C for up to 2 weeks prior to imaging or image immediately.
NOTE: Minor optimizations may be needed depending on the antibody to ensure proper staining. The concentration (1:200) and time (24 h) are significantly higher than in typical 2D monolayer cell culture because 3D staining requires diffusion through the hydrogel and the spheroids.
- After staining the spheroids, clear the spheroid to improve transparency for imaging by replacing PBS with a sequential concentration increase of formamide (optional).
- Aspirate the 1x PBS from each well. Add 500 µL of 20% (v/v) formamide to each well and allow the hydrogel to incubate for 90 min. Aspirate the formamide using a 1000 µL pipette and collect the waste in a waste container.
- Add 500 µL of 40% (v/v) formamide to the well. Allow the hydrogel to incubate in the solution for 90 min. Aspirate the formamide and collect the waste in the waste container.
- Add 500 µL of 80% v/v formamide to each well and incubate for 90 min. Aspirate the formamide and dispose in the waste container. Add 500 µL of 100% (v/v) of formamide and allow 24 h incubation prior to imaging. When clearing is finished, properly dispose of formamide waste through the appropriate services of a laboratory waste management system.
NOTE: Clearing allows for confocal imaging into the core of the spheroid and is optional if only the periphery of the spheroid is being investigated.
- Imaging hydrogel-encapsulated spheroids using confocal microscopy.
NOTE: Any microscope – inverted, fluorescent, or confocal – can be used for cell imaging; however, confocal allows for the isolation of single planes.- Place the hydrogels in chambered wells with glass coverslip bottoms and position the spheroids as close to the coverslip as possible.
NOTE: Glass coverslips or chambered wells with glass coverslip bottoms can be used. It is crucial to keep hydrogels hydrated as dehydrated samples will result in poor imaging quality. - Image the samples with a long-working distance objective (10x-20x) to allow for imaging deep into the spheroid using Z-stacks for 3D reconstructions.
NOTE: Higher magnification objectives allow more detailed imaging and optical sectioning but sacrifice image depth. - Quantify the amount of signal present in the optical section relative to the total area of the spheroid for both the cleared and uncleared signal using equation 2.
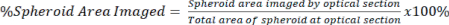 Eq. 2
Eq. 2
- Place the hydrogels in chambered wells with glass coverslip bottoms and position the spheroids as close to the coverslip as possible.
Spheroid-based drug screening platforms to study chemotherapeutic effects are increasingly sought after due to the emphasis on modulating the tumor microenvironment upon spheroid encapsulation in biomaterials replicating native tissue. Here we developed a method for multicellular tumor spheroid preparation and subsequent encapsulation and imaging in a 3D hydrogel. The spheroids are prepared in microwell molds (Figure 3A,B), which result in spheroids with spherical shapes and tightly controlled polydispersity. For example, for spheroids with 3,300 cells per microwell, the average spheroid size was ~250 µm, and circularity was >0.8, where 1 is a perfect sphere (Figure 3C,D). The percent coefficient of variance (%CV) for spheroid diameter was 19.3%, and %CV for circularity was 4.5%. Spheroid diameters were dependent on the number of cells per microwell, as shown in Table 1. Note that some cells in the microwells might not be incorporated in the spheroid and will be washed off during the spheroid harvesting step.
The spheroid is able to be imaged at varying Z-stack depths, allowing for cell viability for each location within the Z-stack (Figure 4). Note that due to confocal imaging limitations and the high cell density, the spheroid core could not be imaged fully. As discussed later, spheroid clearing or sectioning might be needed for improved imaging throughout the spheroid. Through this method, the spheroid viability was determined to be high (~90%) immediately post-harvest and encapsulation, even though cell viability dipped slightly to 85% in the spheroid core compared to the periphery. To ensure viability throughout the spheroid, spheroids were dissociated using acutase, and the viability of the dissociated cells was calculated using the same method and found to be equally high (>90%). A max projection utilizing the spheroid stack represents the highest point of light intensity within each location of the Z-stack compressed into one image (Figure 4B).
Representative images of free-floating spheroids (no gel) and encapsulated spheroids (PEG gel) stained for stemness markers Nestin and SOX2 are shown in Figure 5. Nestin is an intermediate filament and a stem cell marker present in gliomas. SOX2 is a transcription factor for self-renewal, also present in gliomas. SOX2 was found to be co-localized with DAPI in the nucleus, while Nestin was present throughout the cells. The data shows no difference between the gel and no gel conditions, possibly due to the PEG gel used here being inert and not facilitating cell-matrix interactions through integrin signaling.
A major limitation of imaging is the high spheroid density, making it difficult to image into the spheroid core. Common methods to image the spheroid core include sectioning and clearing the sections. This can work well with free-floating spheroids, but sectioning hydrogels is difficult, and most tissue clearing involves dehydrating samples, which results in deformed samples. Here we adapted a protocol from Kuwajima et al.28 for hydrogel samples to maintain the spheroid structure while still clearing the spheroids. To demonstrate the utility of the clearing method, spheroids were fixed, stained with PI, and cleared (Figure 6). Figure 6A shows representative images of optical sections at ~90 µm into cleared and uncleared spheroids and the total area of the spheroid when the spheroid area is filled. When the clearing was performed, the spheroid core could be imaged ~30 µm deeper, compared to an uncleared spheroid (Figure 6B). Clearing can be very beneficial for immunofluorescence, as the spatial organization of biomarkers can be analyzed into the core of spheroids. This method can only be used for fixed samples as the membrane integrity is disrupted.
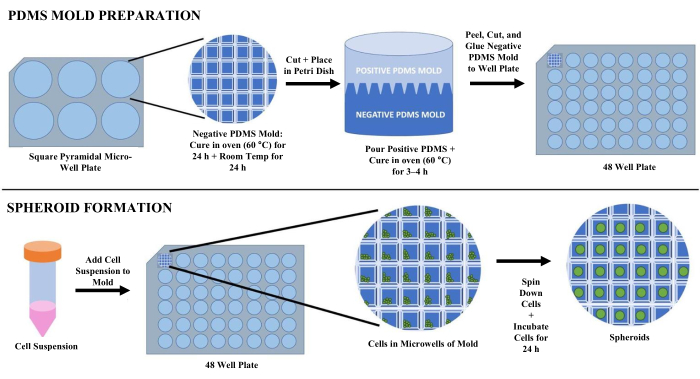
Figure 1: PDMS mold fabrication and spheroid formation. PDMS precursor solutions are added to square pyramidal microwell plates to form PDMS molds. The dissociated cell suspension is added to the formed PMDS molds and spun down to allow for spheroid formation after 24 h. Please click here to view a larger version of this figure.
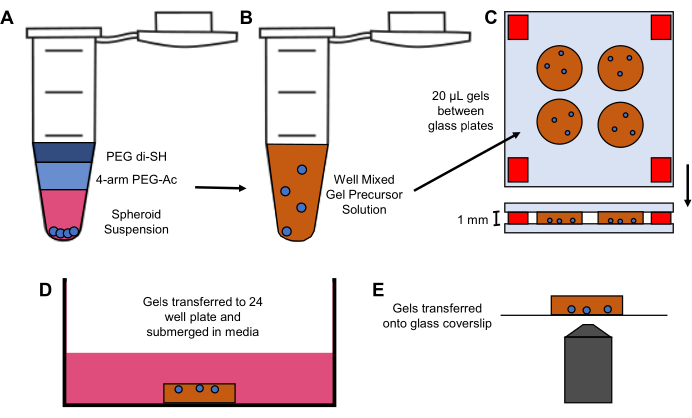
Figure 2: Hydrogel encapsulation, culturing, and imaging. (A) 4-arm PEG-Ac, PEG diSH, and the spheroid suspension is combined in a microcentrifuge tube and (B) mixed well by pipetting up and down to form a gel precursor solution. (C) 20 µL of the gel precursor solution is pipetted onto a glass slide, and a second glass slide is placed on top of the solution and separated by 1 mm spacers. (D) Hydrogel is transferred to 24 well plates with spheroids facing up and is covered with media (500 µL). (E) Hydrogel is transferred to a glass coverslip for microscopy imaging. Please click here to view a larger version of this figure.
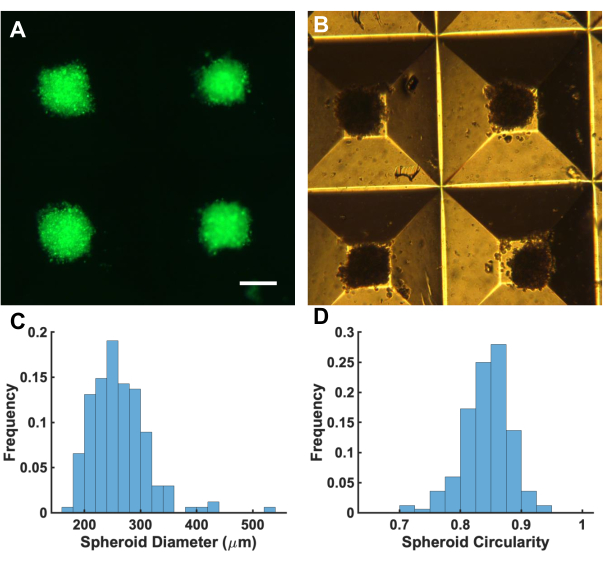
Figure 3: Spheroid formation in microwells prior to harvest, 24 h after initial cell seeding in the microwells. (A) Spheroids in microwell stained with DiOC (Green). Scale bar is 200 µm. (B) Brightfield image of spheroids in microwells. (C) Histogram of spheroid diameter in microwells. (D) Histogram of spheroid circularity in microwells. Please click here to view a larger version of this figure.
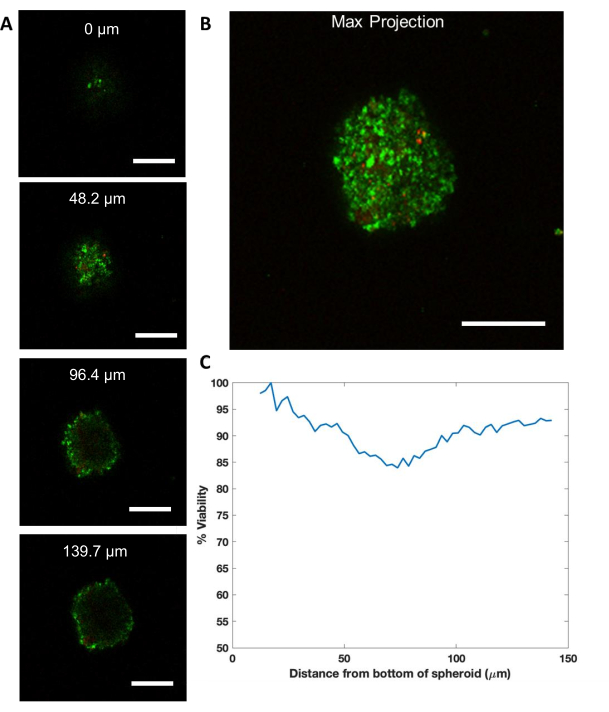
Figure 4: Representative Z-stack confocal image of live spheroid for live dead analysis. (A) Images from 4 slices of Z-stack with DiOC (green) and PI (red). Scale bar is 200 µm. (B) Max projection of Z-Stack. (C) Cell viability as a function of spheroid depth. Please click here to view a larger version of this figure.
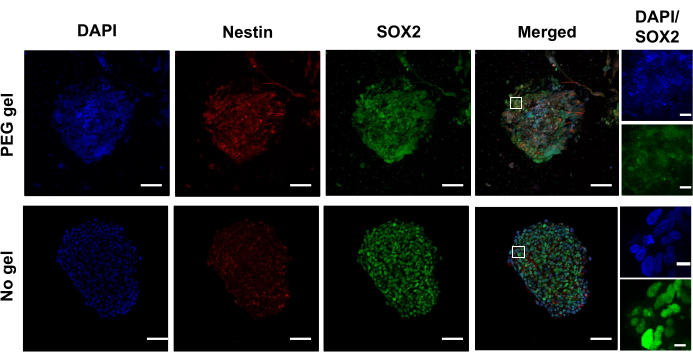
Figure 5: Nestin and SOX2 immunostaining of free-floating (no gel) and hydrogel-encapsulated (PEG gel) U87 cell spheroids at day 5 of culture. Representative confocal images of the spheroids confirm Nestin (red) and SOX2 (green) expression, which were counterstained with DAPI (blue; nucleus). Scale bar is 100 µm. Images cropped and zoomed from the white square show the relationship between the nucleus (blue) and SOX2 (green). Please click here to view a larger version of this figure.
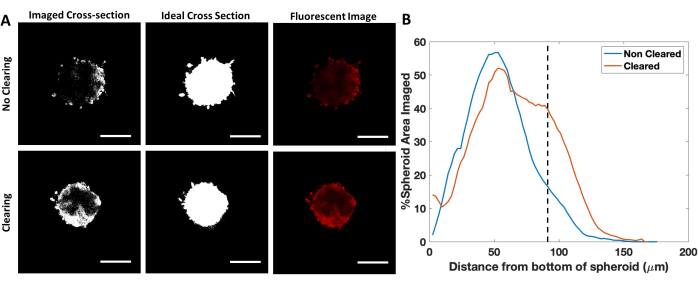
Figure 6: Imaging depth of cleared spheroids. (A) Representative images of spheroids imaged at 91.6 µm into the spheroid and total spheroid area. (B) Percentage of the spheroid area imaged as depth into spheroid increases. Please click here to view a larger version of this figure.
| Microwell Size (µm) | Number of Cells per Spheroid | Number of Cells per Well | Cell Concentration (cells/mL) | Spheroid Diameter (µm) |
| 400 | 200 | 120000 | 240000 | 115.4 ± 13.5 |
| 500 | 300000 | 600000 | 144.6 ± 14.3 | |
| 1000 | 600000 | 1200000 | 176.5 ± 12.5 | |
| 800 | 2000 | 300000 | 600000 | 212.4 ± 15.7 |
| 3000 | 450000 | 900000 | 258.9 ± 16.3 | |
| 4000 | 600000 | 1200000 | 305.7 ± 21.6 | |
| 5000 | 750000 | 1500000 | 323.4 ± 29.8 |
Table 1: Speroid diameter and microwell size. The calculated number of cells per spheroid, number of cells per well of a 48-well plate, cell concentration, and resulting spheroid diameter depending on the microwell size of the negative PDMS mold.
