This paper provides a reliable assay to report the relative cell surface expression of membrane proteins such as ion channels expressed in recombinant cells using the existing flow cytometry technology. Ion channels are pore-forming membrane proteins that are responsible for controlling electrical signals by gating the flow of ions across the cell membrane. They are classified by the activation mechanism, nature, and selectivity of ion species transiting through the pore where they are localized. At the cellular and tissue levels, the macroscopic ion fluxes through ion channels are the product of biophysical (gating and permeation), biochemical (phosphorylation), and biogenesis (synthesis, glycosylation, trafficking, and degradation) properties1. Each of these processes is unique to every type of ion channels and is optimized to fulfill the physiological role of the ion channel. Consequently, alterations in any of these fine-tuned processes through an inherited or a genetic modification, often referred to as "channelopathy", can be detrimental to cell homeostasis. It is important to stress that delivering the "right" amount of ion channels at the cell surface is critical to cell homeostasis. Even small increases (gain-of-function) and small decreases (loss-of-function) in ion channel activity have the potential to cause a serious pathology over a lifetime. Defects in the cell surface delivery of mature ion channels is an important determinant in numerous channelopathies, such as cystic fibrosis (CFTR ion channel)2 and cardiac arrhythmias of the long QT syndrome form (cardiac potassium channels)3.
Channelopathies are associated with cardiac sudden death4. The current worldwide prevalence of all cardiac channelopathies is thought to be at least 1:2,000-1:3,000 per individual5 and are responsible for about half of sudden arrhythmic cardiac death cases6. Dysfunction in cardiac voltage-gated sodium-, potassium-, and calcium- selective ion channels are known to play a key role in this process. The L-type CaV1.2 voltage-gated calcium channel is required to initiate synchronized heart muscle contraction. The cardiac L-type CaV1.2 channel is a multi-subunit protein complex composed of the main pore-forming CaVα1 subunit and CaVß and CaVα2δ1 auxiliary subunits7-12. Note that the full complement of auxiliary subunits is required to produce functional CaV1.2 channels at the plasma membrane and dynamic interactions between these subunits are essential to support the normal electric function of the heart13. CaVß promotes the cell surface expression of CaV1.2 channels through a non-covalent nanomolar hydrophobic interaction14. Co-expression of the CaVα2δ1 subunit with CaVß-bound CaVα1 stimulates peak current expression (5 to 10-fold) and promotes channel activation at more negative voltages. Gain-of-function mutations of the pore-forming subunit CaV1.2 have been associated with a form of ventricular arrhythmias called the long QT syndrome15 whereas a host of point mutations in the three main subunits forming the L-type CaV1.2 channel have been identified in subjects suffering from arrhythmias of the short QT syndrome form16,17. Ion channels are membrane proteins that can be investigated from a biochemical perspective (protein chemistry) or using electrophysiological tools (current-generating machines) and often using these complementary approaches. Electrophysiology, in particular whole-cell patch-clamping, is a suitable approach to elucidate the function of ion channels15 but cannot resolve modifications in protein trafficking from changes in their biophysical properties. Protein chemistry has, however, often limited use due to the relatively low expression of large membrane proteins relative to smaller soluble proteins. Robust high-throughput methods using fluorescence readout need to be developed in order to specifically address defects in protein biogenesis causing changes in the cell surface expression of ion channels.
Flow cytometry is a biophysical technology employed in cell counting, sorting, biomarker detection, and protein engineering18. When a sample solution of live cells or particles is injected into a flow cytometer, the cells are ordered into a single stream that can be probed by the machine's detection system (Figure 1). The first flow cytometer instrument produced in 195619 detected only one parameter but modern flow cytometers have multiple lasers and fluorescence detectors that allow the detection of more than 30 fluorescent parameters20,21. Filters and mirrors (emission optics) direct the light scatter or fluorescent light of cells to an electronic network (photodiode and detectors) that convert the light proportionally to its intensity. Digital data are analyzed using specialized software and the primary output is displayed as a dot plot21.
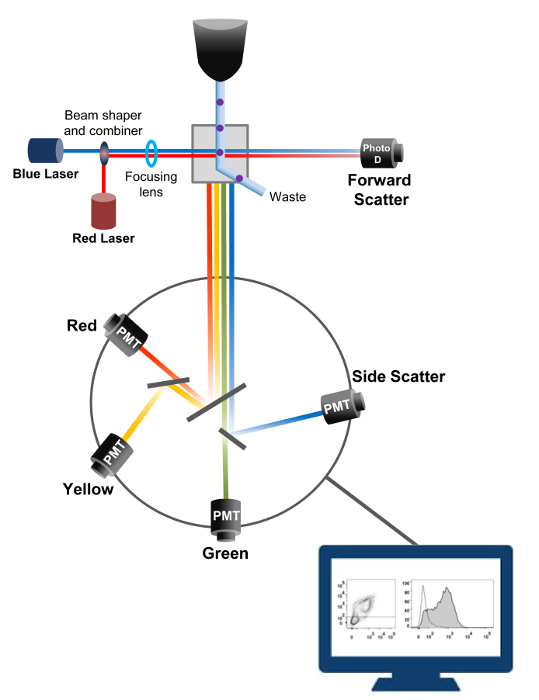
Figure 1: Biophysical principles of flow cytometry sorting. Single cells are pushed through a nozzle under high pressure within a stream of sheath fluid which moves them across one or more laser interrogation points. The light beam is deflected by the passing cells and the light collected in the forward direction (Forward Scatter, FCS) is sent to a photodiode that converts the light into a signal proportional to the size of the cell. The light is also collected at a 90° angle to the laser path and sent to detectors (also called photomultipliers (PMT)). This light is routed through dichroic mirrors that permit the detection of the side scatter signal (SSC), which reflects the granularity within the cells, and the fluorescent emissions if excited fluorochromes are present in the cell. Three detectors (Green, Yellow, and Red) are represented with different wavelength bandpass filters, allowing the simultaneous detection of different fluorochromes. The different signals are digitized by an external computer and converted into data that will be analyzed to quantify the characteristics of the cells. Please click here to view a larger version of this figure.
The high-throughput capacity of flow cytometers was exploited to quantify the relative membrane expression of recombinant wild-type and trafficking-deficient voltage-gated L-type CaV1.2 channels and associated subunits in live cells. cDNA constructs coding for the proteins were doubly tagged to simultaneously carry an extracellular non-fluorescent epitope that can be detected by an impermeable fluorescent conjugated antibody and an intracellular fluorophore that is constitutively fluorescent. Both the extracellular epitope, inserted in an extracellular loop of the protein, and the intracellular fluorophore, inserted after the C-terminus, are translated with the protein. In this series of experiments, the CaVα2δ1 protein was engineered to express an extracellular hemagglutinin (HA) epitope (YPYDVPDYA) detected by an impermeable FITC (Fluorescein isothiocyanate)-conjugated anti-HA and mCherry as the intrinsic intracellular fluorophore. To determine the relative cell surface expression level of the mCherry-CaVα2δ1 HA-tagged protein, recombinant cells expressing the fusion protein were harvested after transfection, and stained with the FITC-conjugated mouse monoclonal anti-HA epitope tag antibody (Figure 2). FITC is an organic fluorescent compound that is considerably smaller than enzyme reporters and therefore not as likely to interfere with biological function. mCherry- CaVα2δ1-HA overexpressed in tsA-201cells, produces a significant 3-log increase in the FITC fluorescence and mCherry fluorescence on two-dimensional plots22. Given that the HA epitope is located in the extracellular portion of the protein, the fluorescence intensity for FITC obtained in the presence of intact cells reflect the relative index of the cell surface expression of HA-tagged protein. The accessibility of the HA epitope in the constructs is systematically validated by measuring the FITC signal after cell permeabilization. This measure also serves to corroborate the normalized total protein expression since the relative fluorescence intensities for FITC estimated in permeabilized cells are qualitatively comparable to the relative fluorescence values for mCherry measured under permeabilized and non-permeabilized conditions22,23. It is important to note that the intrinsic fluorescence spectrum is shifted toward higher values after permeabilization but that the only value being reported is the change in fluorescence intensity as compared to the control construct. Relative changes in the fluorescence intensity for the test constructs are estimated using the ΔMean Fluorescence Intensity (ΔMFI) values for each fluorophore (mCherry or FITC). Experiments are designed to measure the fluorescence intensity of the test construct relative to the fluorescence intensity of the control construct expressed under the same conditions to limit experimental variations in the intrinsic fluorescence of the fluorophore-conjugated antibody. Two membrane proteins were successfully studied using this assay: the pore-forming subunit of the L-type voltage-gated calcium channel CaV1.214,22 and in a different series of experiments, the extracellular auxiliary CaVα2δ1 subunit22,23. The following protocol was used to determine the cell surface expression of the CaVα2δ1 subunit of the cardiac L-type CaV1.2 channel under control conditions and after mutations affecting the posttranslational modification of the ion channel. Under standardized experimental conditions, the cell surface fluorescence of FITC increases quasi-linearly with the expression of cDNA coding for the mCherry-CaVα2δ1-HA proteins (Figure 5 from reference22).
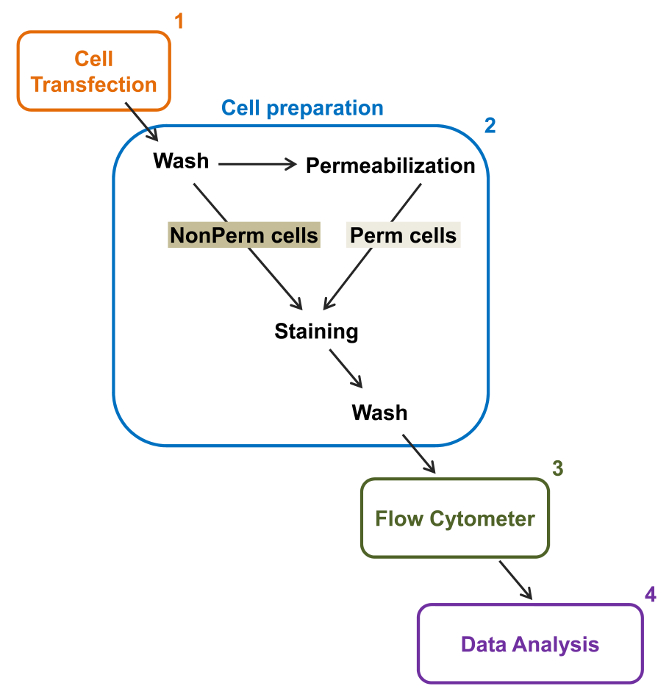
Figure 2: Schematic representation of total and membrane labeling in the flow cytometry experimental protocol. The scheme outlines some of the main steps necessary to quantify the relative total and cell surface expression of recombinant ion channels by flow cytometry. Cells are transfected with the double-tagged construction mCherry-CaVα2δ1-HA in tsA-201 cells (1) and stained before or after permeabilization (2). Multiparameter data are acquired in a flow cytometer (3) for multivariate analysis (4). Please click here to view a larger version of this figure.
