To determine if the procedure described above is successful, the embryos can be observed during the cleavage stages to confirm the appearance of the stereotypical early-embryonic cleavage pattern18, as well as at 24 h post-fertilization (hpf) to confirm the proper development of the basic body plan. This procedure allows for the manipulation of maternal products for functional studies through the injection of reagents, such as mRNAs and MOs, during oocyte development.
Manipulation of maternal products for functional studies:
mRNA coding for wild-type and mutated products can be injected into in vitro maturing oocytes to test the effect of the manipulation of maternal gene function during meiosis and the early embryonic stages. For example, wild-type embryos can be injected with wild-type mRNA to test for the effect of product overexpression, or with mutant RNA to test for potential dominant (e.g., gain-of-function and antimorphic) effects in these processes. Oocytes in in vitro culture are able to produce protein from exogenous mRNA throughout oocyte development, as shown by the expression of GFP from injected mRNA, although only oocytes that initiate culture conditions at stage IV of oogenesis can develop into mature stage V oocytes (Figure 2B-2D, see also Nair et al.3). The expression of wild-type product through injected mRNA is also instrumental for the rescue of maternal-effect mutations to confirm gene identity during positional cloning3,24. In this case, wild-type mRNA is injected into oocytes from homozygous mutant females to test whether wild-type products can rescue the mutant phenotype. This genetic rescue is illustrated in Figure 3A-3C, where injected AurB mRNA is shown to rescue the phenotypic effects of a mutation in its corresponding gene, cellular island (cei) (Figure 3A-3C). The embryos from a control group are also allowed to develop to show the corresponding mutant phenotype3. Mutant RNA can also be injected into mutant oocytes to test whether the mutated product retains partial function by comparing the extent of rescue to that caused by the wild-type product.
Protein expression from mRNA injected into developing oocytes appears to occur with little or no delay. Strong GFP expression is observed within 2 h of the injection of the corresponding mRNA, regardless of the developmental stage of the oocyte3 (Figure 2B-2D; see also Nair et al.3). Products such as mCherry:Sas6 and Birc5b(Motley):GFP can be observed immediately after fertilization and during the first embryonic cell cycle3,25. The injection of mRNA during oogenesis also leads to the production of protein that, regardless of the fused tagged moiety, is functional immediately after fertilization, as shown in the case of maternal products for cellular island/aurB3, futile cycle/lrmp24, and motley/bir5b25. Translation-blocking MOs also have an effect immediately after fertilization, as shown for futile cycle/Lrmp3 and at later stages of embryogenesis, as in the case of mission impossible/dhx163. Splice-blocking MOs, when injected in maturing oocytes, may not have an effect on maternal function, likely due to already-present mature maternal transcripts in stage IV oocytes3.
Generation of morphants:
When using a MO corresponding to a gene with an already-identified mutation, the morphant phenotype is expected to mimic the mutant phenotype. Morphants are compared to uninjected embryos and to those injected with a standard, control MO. The injection of a translation-blocking morpholino can successfully phenocopy the known mutant phenotype3, as shown by the injection of Lrmp MO into oocytes, which mimics the mutant phenotype of the corresponding mutation, futile cycle (Figure 3D-F). The specificity of MOs can be determined through the same approaches used when injecting MOs into embryos (reviewed previously)19,20.
Expression of fluorescently labeled fusion proteins:
mRNA coding for gene products of interest fused to fluorescent proteins (e.g., GFP and mCherry) can also be injected into either wild-type or mutant oocytes to visualize the corresponding products within the early embryo (i.e., reflecting a subcellular localization pattern; Figure 3G and 3H). mRNAs coding for similar fusions but involving the mutant allele can be similarly expressed to test whether the mutation affects any potential subcellular localizations.
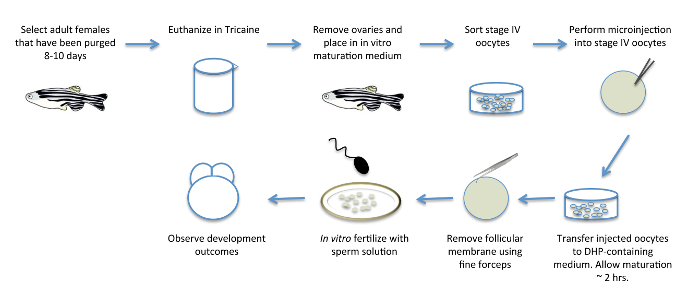
Figure 1: In Vitro Oocyte Maturation. Schematic diagram showing the various steps involved in in vitro oocyte maturation. Adult females are pre-selected for egg-laying 8 days prior to the procedure, to purge them of eggs that have already matured and to promote new oocyte development. The ovaries, containing developing oocytes, are removed from females and transferred to a medium containing the hormone DHP to induce oocyte maturation in vitro. After removal from females and immediately prior to or during oocyte maturation, oocytes can be injected with RNA products and other reagents for the functional manipulation of maternal factors. Mature oocytes are manually defolliculated and fertilized in vitro with a sperm solution to yield fertilized, viable embryos. Please click here to view a larger version of this figure.
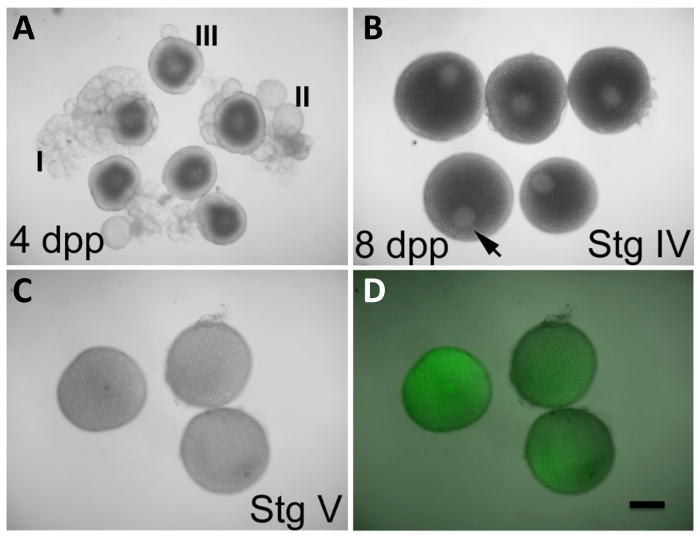
Figure 2: In Vitro Oocyte Maturation and Expression of Products from Injected mRNA. (A) Oocytes at stages I-III, observed in ovaries from females 4 days post-purging (dpp). (B) Oocytes at stage IV, observed in ovaries from females 8 dpp. The germinal vesicle (GV, arrowhead) is clearly visible and occupies an eccentric position. The stage III oocytes in (A) also exhibit a readily apparent GV, but it is found centered in the oocyte. Stage IV oocytes in (B) have a size that is near maximal compared to that of mature oocytes in (C). (C and D) Oocytes at stage V (mature oocytes) after 2 h in in vitro maturation conditions initiated at stage IV, as in (B), and injected during maturation with GFP-encoding, in vitro transcribed mRNA (C, visible light only; D, overlay of visible light and GFP fluorescence). The GV is no longer apparent in stage V oocytes, which are also less opaque than stage IV oocytes. Injected oocytes express GFP protein (D). Defolliculation of mature stage IV oocytes is described in the protocol section. Scale bar = 300 µm. All panels were reproduced, with permission, from Nair et al3. Please click here to view a larger version of this figure.
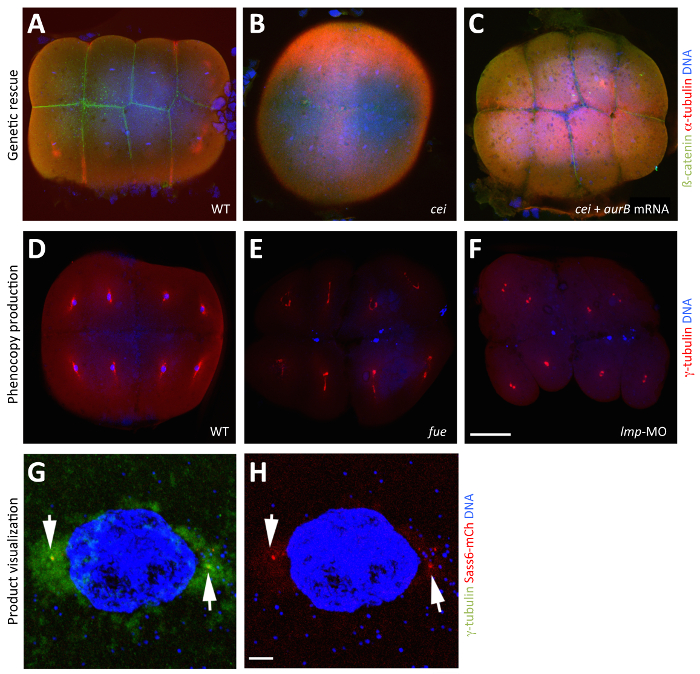
Figure 3: Manipulation and Visualization of Maternal Products through In Vitro Oocyte Maturation. (A–C) Rescue of the maternal-effect phenotype caused by a mutation in cellular island/aurora B through the injection of wild-type aurB mRNA into stage IV cei/aurB oocytes. Merged images showing animal views of 65 mpf fixed blastodiscs, visualized with anti-ß-catenin antibodies to highlight membranes (green), anti-α-tubulin antibodies to indicate microtubules (red), and DAPI to designate DNA (blue). (A) β-catenin accumulation in wild-type embryos, indicative of normal furrow maturation. (B) A cei/aurB embryo from an uninjected cei/aurB stage IV oocyte shows partial, rudimentary furrows that do not accumulate β-catenin. (C) A rescued cei/aurB embryo from a cei/aurB oocyte injected with wild-type aurB mRNA showing robust β-catenin accumulation at the furrows. (D–F) Phenocopy of the maternal-effect caused by a mutation in futile cycle/lrmp from the injection of Lrmp morpholino into stage IV oocytes. Merged images showing animal views of 70 mpf fixed blastodiscs, visualized with anti-γ-tubulin antibodies to indicate centrosomes (red) and DAPI to designate DNA (blue). (D) In wild-type embryos, each nucleus associates with γ-tubulin, a marker for centrosomal material. (E) In maternal-effect fue mutants, pronuclear fusion fails, resulting in two to three patches of DNA labels corresponding to unfused parental pro-nuclei and the polar body for meiosis II. (F) In Lrmp morphants, where maternal Lrmp function is inhibited, the nuclei similarly fail to divide and, in addition, fail to associate with γ-tubulin. (G and H) Visualization of maternally produced product in the early embryo. Sass6-mCherry protein from exogenous mRNA injected into stage IV oocytes localizes to the centrioles. Merged images showing animal views of 40 mpf fixed blastodiscs, visualized with anti-γ-tubulin antibodies to highlight centrosomes (green), mCherry fluorescence to indicate Sass6 (red), and DAPI to designate DNA (blue). (G) Expressed Sass6-mCherry protein localizes to foci labeled by the centrosome marker γ-tubulin at sites flanking the nucleus (arrows). (H) The same image, showing only Sass6-mCherry and DAPI for clarity. The embryos in (G and H) are fixed, but the fluorescence of expressed products, such as mCherry fusions, can also be observed in live embryos (not shown). Scale bar = 100 µm in A-F and 10 µm in G and H. All panels were reproduced, with permission, from Nair et al3. Please click here to view a larger version of this figure.
| Stages of Oogenesis | Oocyte diameter (μm) | Key landmark(s) |
| 1A – Prefollicle | 7 to 20 | Initiation of active transcription, accumulation of nucleoli |
| 1B – Follicle | 20 to 140 | Decondensation of chromosomes |
| II – Cortical alveolus | 140 to 340 | Cortical alveoli production |
| III – Vitellogenesis | 340 to 690 | Darkening of ooplasm by accumulation of yolk precursor protein and lipids |
| Early IV – Oocyte maturation | 690 to 730 (lower range) | Asymmetric localization of germinal vesicle |
| Early IV – Oocyte maturation | 690 to 730 (upper range) | Germinal vesicle disappears, arrest at metaphase II |
| V – Mature oocyte | ~750 | Ooplasm/yolk becomes translucent |
Table 1: Landmarks of Oocyte Development in Zebrafish.
