The workflow for sample preparation and MS analysis is depicted in Figure 1. After the dissection of neuronal tissue, homogenization, extraction, and desalting are performed to purify neuropeptide samples. If isotopic label-based quantification is desired, samples are then labeled and desalted once again. The resulting sample is analyzed through LC-MS/MS for neuropeptide identification and quantification.
Neuropeptides identified through the proteomics software should have good peptide fragmentation sequence coverage, however, this is not globally defined or standardized. For absolute identification, every amino acid should produce a fragment ion that provides unambiguous identification and localization. This must also be compared with a synthesized peptide for confirmation of the intensity of each fragment ion. As the cost for performing this on every putative identification is not feasible, identification is commonly described based on confidence where more observed fragment ions increase peptide identification confidence. While Figure 2A,B depicts two neuropeptides that were both identified with 100% sequence coverage and a low mass error as defined by the max limit of 0.02 Da, the poor fragmentation coverage (from only three ions) observed only for the neuropeptide in Figure 2B decreases the confidence of identification of a specific isoform. Figure 2C depicts the extracted ion chromatograms (XICs), which is a plot containing the signal intensity of a selected m/z value as a function of retention time, of a neuropeptide detected in two samples used for LFQ. The retention times for the neuropeptide differ slightly because it was identified in two separate and consecutive runs; however, the difference is within the reliable threshold value of 1 min. Thus, the ratio between the software-calculated area under the curve from the XIC is used for the LFQ of this neuropeptide.
For the quantitation of dimethyl labeled neuropeptides, the MS1 spectrum should contain a peak at the theoretical neuropeptide m/z value and a peak at the m/z value with a mass shift that correlates to the mass difference between the isotopic labeling reagents used. In Figure 2D, the mass shift of this 2-plex dimethyl labeled sample is 4.025 Da. The area under the curve of the precursor ion from its XICs is then calculated by the software and used to calculate relative abundance ratios. A simplified version of the proteomics software export table containing identified neuropeptides and their LFQ ratios is shown in Table 1. Similar results are obtained for isotopically labeled neuropeptides.
Software algorithms enable the de novo sequencing of spectra to detect novel putative neuropeptides. When claiming the detection of putative novel neuropeptides, high confidence identifications are ideal cases where all amino acids are identified and localized unambiguously, based on fragment ion observation. Figure 3 depicts the spectrum of a de novo sequenced peptide containing the -RYamide motif at the C-terminal, a conserved sequence motif shared by known neuropeptides of the crustacean RYamide family22. A peptide was matched from the database with 100% sequence coverage, all amino acid forming fragment ions observed, low fragment ion mass error as defined by the max limit of 0.02 Da and contained a gaussian elution profile. These results indicate that an endogenous peptide belonging to the crustacean -RYamide neuropeptide family was observed.
MALDI-MS spot measurements can provide neuropeptide identifications that are complementary to LC-ESI-MS identification, as well as offer higher throughput capabilities. After crude tissue homogenate is extracted for neuropeptides, desalted, and labeled (if desired), the sample can be mixed with matrix and spotted on the MALDI stainless steel target plate, as shown in Figure 4A. Successful pipetting of homogenous sample spots produces clearly resolved peaks, especially within the calibration spectrum (Figure 4B). When using a MALDI-TOF instrument, the instrument must be calibrated at the beginning of each experiment. Any analytes with known masses can be used to calibrate the instrument if it is within the desired mass range of the sample. Here, red phosphorus is used for the positive ion mass calibration of the instrument. It has advantages over using peptide calibration mixes due to its stability at room temperature, cheap cost, abundant peaks due to its polymerization, high signal-to-noise ratio, and it does not require a matrix for ionization.
For MALDI-MS imaging of neuropeptides, the MALDI TOF/TOF instrument used requires manual image calibration of the ITO-coated slide (step 3.1.10) to correlate the optical image with the sample. The diagram in Figure 5 shows the proper placement of the whiteout crosshairs to be used as teach points to allow the instrument to correlate the scanned optical image with the actual sample plate. It also illustrates areas of the ITO-coated slide that should be avoided by the user (i.e., do not contain sample or matrix). For mass calibration, the placement of the calibration sample spot relative to the tissue sample on the ITO-coated slide directly impacts the mass error due to the inherent nature of time-of-flight mass analyzers, although the magnitude of this issue is dependent on the abundance of the target analytes. A solution to this problem is to manually check peaks from the MS1 spectra from different tissue sections for evidence of peak shifting. If there is peak shifting, consider adding additional mass calibration spots onto the ITO-coated slide right next to each tissue section. From there, the user can average the spectra from the calibration spots together or perform MS imaging on one tissue section at a time using only the calibration spot closest to the tissue section. After the sample is collected, verify that the signal from m/z values corresponding to neuropeptides is only localized within the tissue region (Figure 6) before assigning a putative neuropeptide identification.
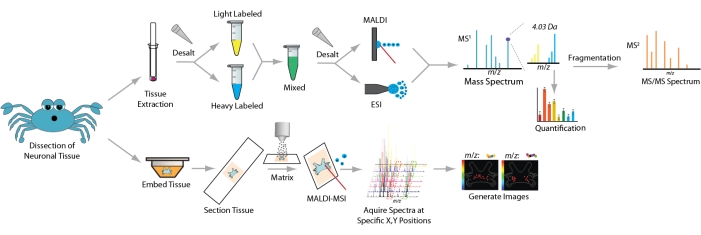
Figure 1: Neuropeptide sample preparation workflow for mass spectrometry analysis. For tissue extract analysis crude tissue homogenate is desalted, labeled with stable isotopic labels, desalted again, and analyzed by MS. For imaging analysis intact tissue is embedded, cryosectioned, applied with matrix, and analyzed by MALDI-MSI. Please click here to view a larger version of this figure.
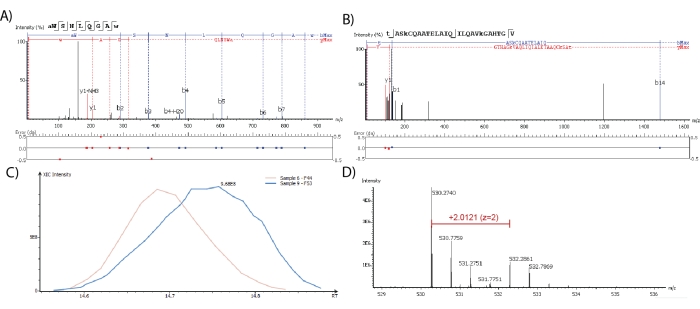
Figure 2: Identification and quantification performed through the proteomics software. Neuropeptides are detected through spectra of ranging quality with (A) good or (B) poor MS2 fragmentation coverage. Fragment ion mass matching error is shown below the spectra. (C) XIC profile shapes and retention time (RT) can be manually inspected for neuropeptides quantified through LFQ. (D) MS1 spectra are used to detect and quantify dimethyl labeled neuropeptides. Please click here to view a larger version of this figure.
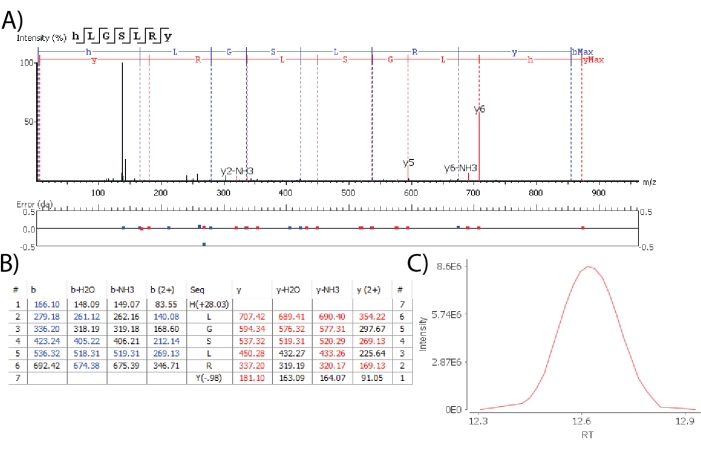
Figure 3: De novo sequencing for novel neuropeptide detection. (A) The MS2 spectrum of a putative novel RYamide demonstrates good fragmentation coverage with low mass error for each fragment. (B) The identified fragment ions are listed for manual inspection. (C) The XIC of the novel neuropeptide is manually inspected for Gaussian peak shape. Abbreviations: RT = retention time. Please click here to view a larger version of this figure.
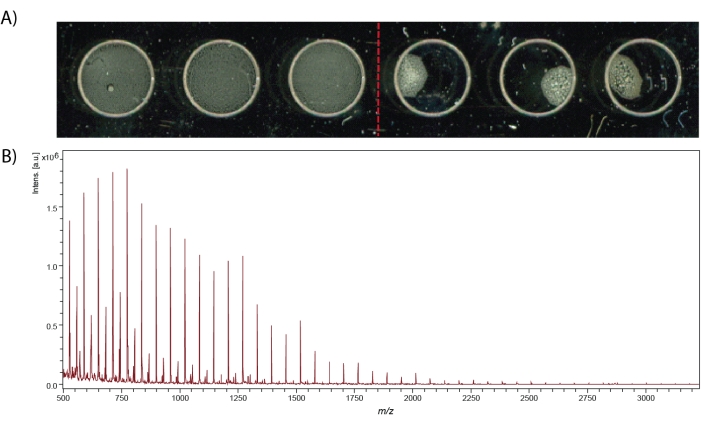
Figure 4: MALDI-MS spots and calibration spectrum. (A) MS spectra quality relies on uniform matrix-peptide distribution in the MALDI stainless steel target well. The left three spots are examples of good spots that touch the edges of the well engraving and the right three spots are examples of bad spots. Both spots contain α-Cyano-4-hydroxycinnamic acid (CHCA) matrix and a peptide standard mix. (B) Calibration spectrum using red phosphorus clusters from 500 – 3200 m/z. Please click here to view a larger version of this figure.
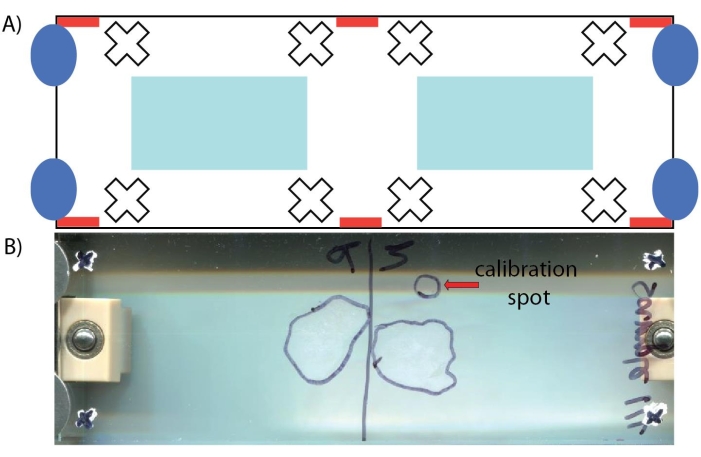
Figure 5: Depiction of ITO-coated glass slide. (A) Schematic with important areas noted: locations to place tissue sections (light blue rectangles), automatic teach points locations that should be avoided (red rectangles), example locations of where teach points may be drawn (white crosshairs), and where screws attach to the adapter plate and should be avoided (dark blue ovals). (B) Photo of glass slide containing two tissue sections, a spot containing a calibration mix, and crosshair marks. The location of the tissue section and calibration spots are outlined on the other side of the glass slide. Please click here to view a larger version of this figure.
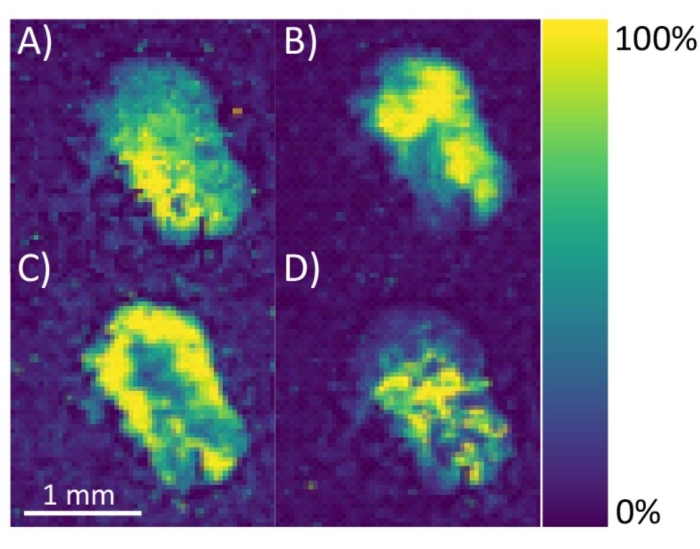
Figure 6: MS images of C. sapidus sinus glands. Neuropeptide [M+H]+ ion distribution images of (A) HL/IGSL/IYRamide (m/z 844.48), (B) Allatostatin A-type NPYAFGLamide or GGPYAFGLamide (m/z 780.40), (C) Allatostatin A-type GQYAFGLamide (m/z 754.39), and (D) RFamide GRNFLRFamide (m/z 908.52) are shown. Images are generated using a ± 50 ppm window from the theoretical m/z value. Color bar indicates the range of signal intensity from 0 to 100%. Please click here to view a larger version of this figure.
Table 1: Database search and LFQ results. Neuropeptides identified and quantified through LC-MS and proteomics software. Identified PTMs are listed along with the intensities of detected peptides in both samples for LFQ, along with the resulting LFQ ratio. The average masses and observed neuropeptide descriptions from the FASTA file are listed. Please click here to download this Table.
