Tonsil and lung adenocarcinoma TMA tissue sections (5 mm thick) were obtained and placed on the middle of gold-coated slides following the specifications regarding tissue size and secure margins of the slides. Free glass margins of 5 mm and 10 mm between the edge of the tissue and the lateral and inferior borders of the glass slides, respectively, are necessary for optimal staining. The tissue sections were baked overnight in an oven prior to staining to assure proper adherence of the section to the slide. The antibody panel was composed of 23 markers. In the same staining batch, a tonsil slide and a TMA slide containing multiple tissues were included to evaluate the expression of markers scantly expressed in lung adenocarcinoma samples (Figure 1). Positive controls need to be chosen according to the targets of the antibodies used in the panels; for example, to evaluate SOX10 expression, melanoma samples are needed, and to evaluate GAFP expression, glioblastoma samples are needed (Figure 2).
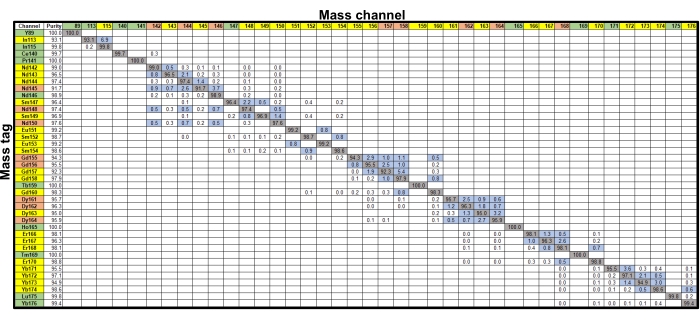
Figure 1: Isotopic purity and channel cross-talk. The matrix shows the percent cross-talk derived from probe purity and oxides (blue boxes, ≥0.5%; clear boxes, <0.5%). For the mass tags, probes that contributed at least 0.5% cross-talk into no channels (green), one or two channels (yellow), or more than two channels (orange) are shown. Mass channels receiving at least 0.5% cross-talk from no probes (green), one or two probes (yellow), or more than two probes (orange) are also shown. Please click here to view a larger version of this figure.
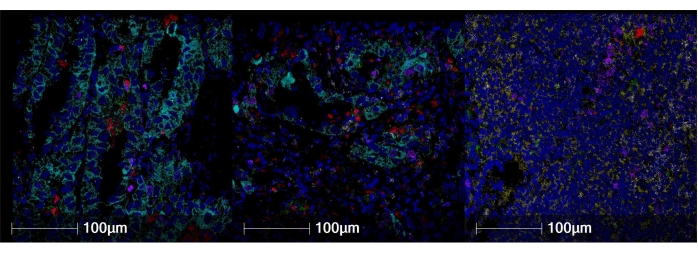
Figure 2: Representative staining images in different tissues. (A) Lung adenocarcinoma. (B) Nonneoplastic kidney. (C) Tonsil. CD20 is shown in yellow, Ki67 is shown in magenta, CD3 is shown in white, CD11c is shown in green, CD68 is shown in red, cytokeratin is shown in cyan, and double-stranded DNA (dsDNA) is shown in blue. Magnification, 200x. Please click here to view a larger version of this figure.
This staining procedure closely resembles standard immunohistochemical staining and occurred on two consecutive days. The five main steps of the staining protocol are 1) excess paraffin removal, 2) antigen retrieval, 3) antibody blocking, 4) antibody staining, and 5) fixation and dehydration (Figure 3). A fundamental step in the staining procedure is to take precautions to avoid metal contamination of the samples, including using metal-free reagents stored in plasticware and careful handling of the samples to limit mechanical damage. Preparing the antibody cocktail immediately before staining is recommended. This reduces the metal exchange. Once the staining was complete, we stored the slides and scanned them the next day. Before image acquisition, a necessary step is slide setup using a web-based image management application (Figure 4).
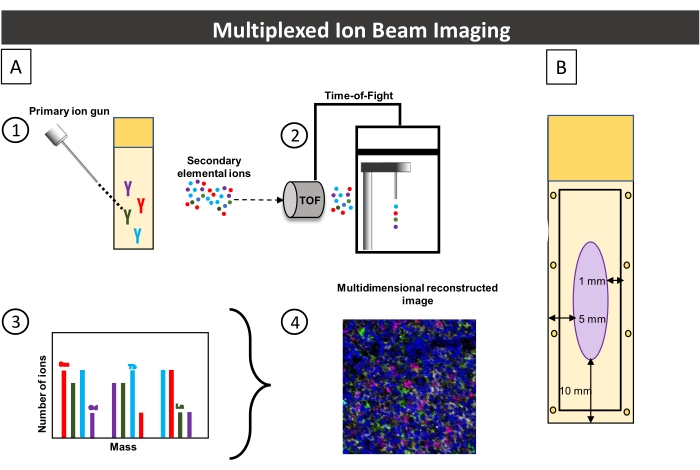
Figure 3: Image acquisition workflow and the associated slide. (A) Image acquisition workflow summary. 1) An FFPE tissue section stained with metal-conjugated antibodies is rasterized using a primary ion gun, releasing secondary ions. 2) A time-of-flight mass spectrometer separates, and measures ions based on their mass at the pixel level. 3) Pixel-based quantification of secondary ions. The y-axis corresponds to the number of ions detected (peak), and the x-axis corresponds to the mass of each metal. 4) Based on the peak of each mass spectrum, multidimensional images are reconstructed. (B) Schematic of the slide used in this procedure. For optimal staining tissue, the section must be positioned at least 5 mm from the slide's lateral edge and 10 mm from its inferior edge. When drawing the hydrophobic barrier around the tissue section, it must be kept inside the rectangle at least 1 mm from the slide's edge. Please click here to view a larger version of this figure.
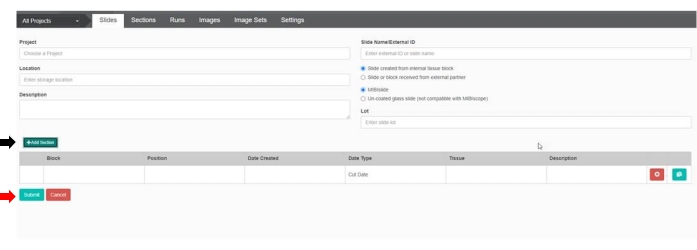
Figure 4: Slide setup in the web-based image management application. Before slide scanning, setting up each slide is necessary. Enter the desired details that can help with slide identification. Click on Add Section (black arrow) to include the block and position information. To save, click Enviar (red arrow). Please click here to view a larger version of this figure.
On the scanning day, turn on the acquisition instrument using the control software. Clicking on Exchange Slide opened the door of the instrument, allowing loading of a slide. We positioned the slide on the loading slot facing up with the label on the right side. Only one slide can be scanned at once. The next step was selecting the FOVs and adjusting the focus and stigmation following the steps described in the protocol. We acquired images by clicking on Start Run. The acquisition time varies depending on the FOV size and resolution desired. The FOV size ranges from 200 μm x 200 μm to 800 μm x 800 μm, which corresponds to a frame size range of 128 pixels x 128 pixels to 2048 pixels x 2048 pixels. Three standard resolutions are available: coarse, fine, and superfine. Scanning larger FOVs at superfine resolution is time-consuming, with the final acquisition time for one FOV ranging from 25 s to 4.7 h (Figure 5).
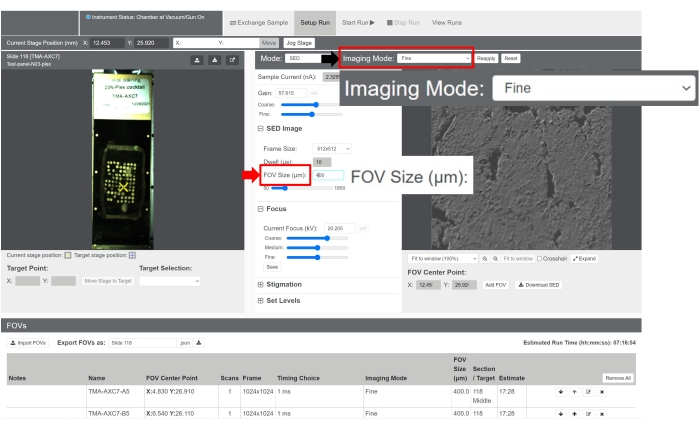
Figure 5: Image acquisition using the control software. The acquisition settings can be adjusted as desired. To modify the scanning resolution, click on the dropdown menu by Imaging Mode (black arrow). To modify the FOV size, enter a number in the FOV Size (μm) field (red arrow). Please click here to view a larger version of this figure.
After scanning was complete, an image set including all FOVs was automatically uploaded to the web-based image management application. Besides storage of images, this application enables the visualization of all channels together or separately, image adjustments, and downloading images for further preparation. The standard visualized image is saved as a TIFF file (Figure 6).
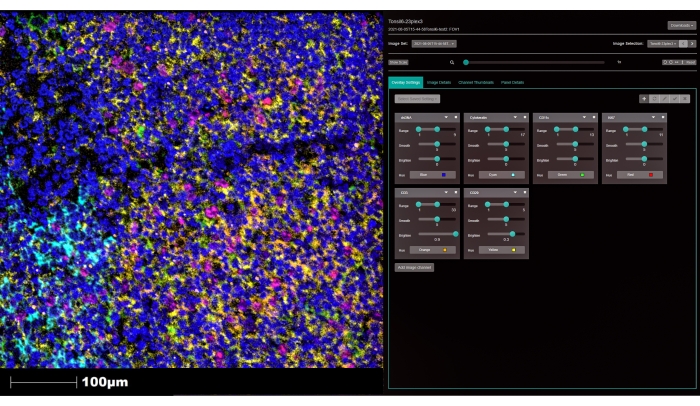
Figure 6: Image visualization using the web-based image management application. Acquired images are stored and visualized using the online platform. Adjusting the visualization parameters and changing the color of each marker is possible. Click on Add image channel (white arrow) to visualize other markers. Magnification, 200x. Please click here to view a larger version of this figure.
Metal interferences are inherent to the metal-labeling methods despite the use of strategies to minimize them. To remove the excess background signals from metal isotope conjugated to the antibodies and prepare images for analysis, we used the associated image processing software (see Table of Materials). The image preparation procedure has two steps: 1) isobaric correction, which removes signals between channels, and 2) filtering, which removes signals caused by aggregates on the image.
To start the isobaric correction, the file containing the saved TIFF image was selected by clicking on the File icon of the input pane. The software automatically loads all archived TIFF images in the file, allowing batch analysis. Next, we corrected the image by clicking on the Filter icon of the input pane. Two resulting files with the suffix -MassCorrected were automatically generated: one archived in TIFF format and the other archived in JSON format. By default, the resulting archives were saved in the same file from which the initial image was loaded (Figure 7).
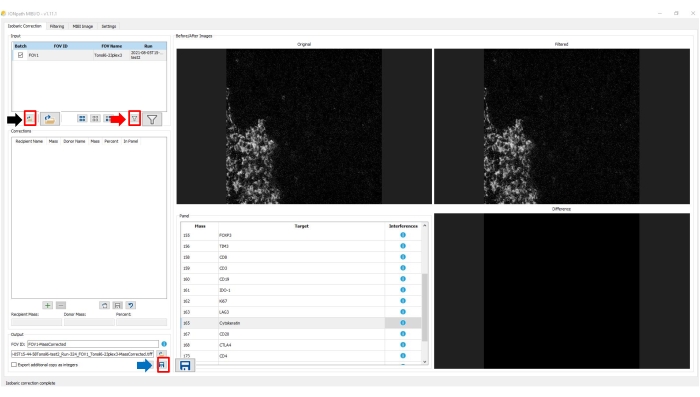
Figure 7: Image preparation: correction step. To load an image for preparation in the image processing software, click on the File icon in the input pane (black arrow) and select the image. To apply the default correction procedure, click on the Filter icon in the input pane (red arrow). To save the updated, corrected image, click on the Floppy disk icon on the output pane. Please click here to view a larger version of this figure.
The second step of image preparation is filtering, which can be selected on the upper tab in the software. We selected the corrected image on the input pane. In this step, automated Voronoi tessellation was used as the filtering parameter. Clicking on the filter icon on the input panel automatically applied the selected filter to all channels. In both image preparation steps (correction and filtering), the images of each channel before and after processing and signal differences are displayed on the right side of the screen.
Similar to the isobaric correction step, two new archives, one in TIFF and one in JSON format, were generated. In this step, the suffix following the file name was -Filtered. Therefore, the final image obtained was named MassCorrected-Filtered.tiff. By completing these steps, we prepared the image for the analysis using the preferred digital pathology software (Figure 8 and Figure 9). By using this technique, we were able to analyze all 23 markers in the antibody panel with minimal interferences between channels at the subcellular level in a single tissue section.
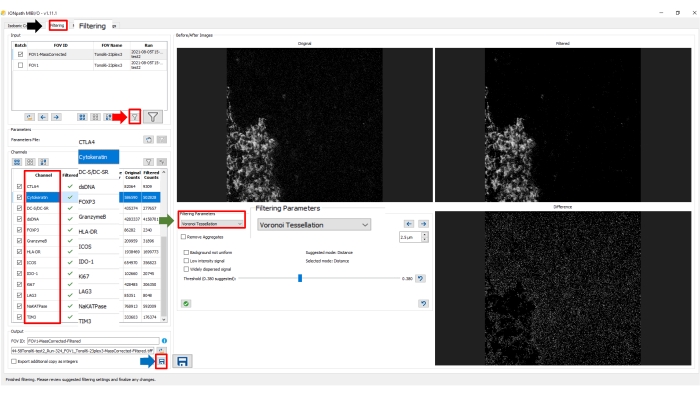
Figure 8: Image preparation: filtering step. Select Filtering in the upper tab (black arrow) in the image processing software. Under Filtering Parameters, select the desired method (green arrow). On the input pane, select MassCorrected image. To apply the selected procedure to the image, click on the Filter icon on the input pane (red arrow). To save the updated filtered image, click on the Floppy disk icon on the output pane. Please click here to view a larger version of this figure.
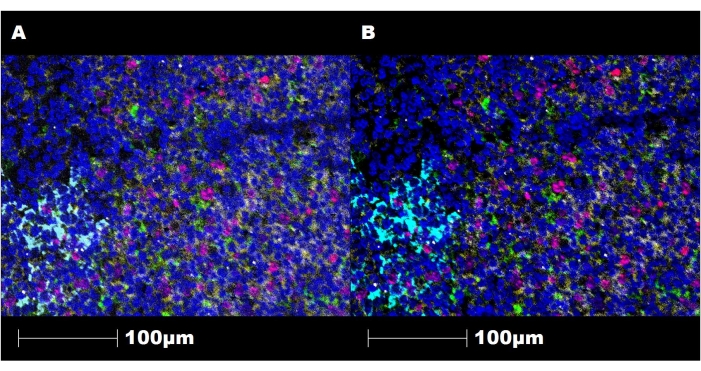
Figure 9: Representative images of the staining before and after image preparation. (A) Image of a tonsil tissue section stained using this technique and visualized using a third-party digital image analysis software program before filtering and correction. (B) Image of the same section under the same conditions after filtering and correction steps. CD20 is shown in yellow, Ki67 is shown in magenta, CD3 is shown in white, CD11c is shown in green, cytokeratin is shown in cyan, and double-stranded DNA (dsDNA) is shown in blue. Magnification, 200x. Please click here to view a larger version of this figure.
Supplementary File 1: Antibody panel list. Each antibody was conjugated to a specific metal isotope with a different mass as listed. Please click here to download this File.
