Analysis of peritoneal macrophage-mediated engulfment of apoptotic thymocytes. Peritoneal macrophages and apoptotic cells were prepared and co-cultured as described in the protocol. Macrophages were detached and stained with PE conjugated anti-CD11b antibody for 20 min on ice. Macrophages were then washed and processed in a flow cytometer. As seen, there is no CFSE positive macrophage in the bottom right quadrant when no apoptotic cells were added into the culture (Figure 1A and Figure 2, first panel). Thioglycollate-stimulated peritoneal macrophages have the variable capacity to engulf apoptotic cells. Up to 30% of macrophages showed positive in the CFSE channel, indicating they have ingested CFSE-labeled apoptotic cells in this experiment (Figure 1). It is worth to note that CFSE positive macrophages spread out in the bottom right quadrant due to different intensities, indicating that the number of apoptotic cells within macrophages is different. Therefore, microscopic observation of macrophage engulfment of apoptotic cells is essential to investigate the capacity of macrophages to ingest apoptotic cells (Figure 2). The higher ratio of apoptotic cells to macrophages not only increases the number of macrophages ingesting apoptotic cells but also enhances the ability of macrophages to ingest more apoptotic cells (Figure 2).
Dose-dependent inhibition of efferocytosis by Mer blockage.
In another set of experiments, we found that about 15% of the macrophages became CFSE positive when CFSE-labeled apoptotic thymocytes (ration of 6:1) were added into the macrophage culture for 4 h, indicating they are the phagocytic macrophages (Figure 3B). One function of Mer on macrophages is to recognize and mediate phagocytosis of apoptotic cells through the bridging molecule, Gas6. To test the percentage of efferocytosis attributed by Mer inhibition, we added anti-Mer antibodies into the culture to block Mer-mediated efferocytosis. Mer antibody blocks macrophage efferocytosis in a dose-dependent manner (Figure 3C, Figure 3D) and the overall blockage may account for about 30% of the efferocytosis efficiency in the current setting (Figure 3), This data was also confirmed in our previous study with Mer knockout macrophages13. We then tested the efficiency of inhibition with the newly FDA approved TAM receptor inhibitor, RXDX-106, which inhibits all TAM receptors with different affinities (Axl>>Tyro3>Mer). RXDX-106 was added into the macrophage culture 2 hours before co-incubation with apoptotic thymocytes. As shown in Figure 4, RXDX-106 inhibited macrophage efferocytosis in a dose dependent manner. The saturated inhibition concentration was about 100 nM (Figure 4, solid line), a concentration that appears to be more effective than Mer-deficiency (Figure 4 dotted line) or Mer antibody (Figure 3, panel D) alone. Since macrophage expresses all three TAM receptors on the surface16, it is expected to see that higher concentrations of RXDX-106 (over 100 nM) will block all three receptors, and therefore, will be more effective in blocking efferocytosis than targeting a single TAM receptor, Mer.
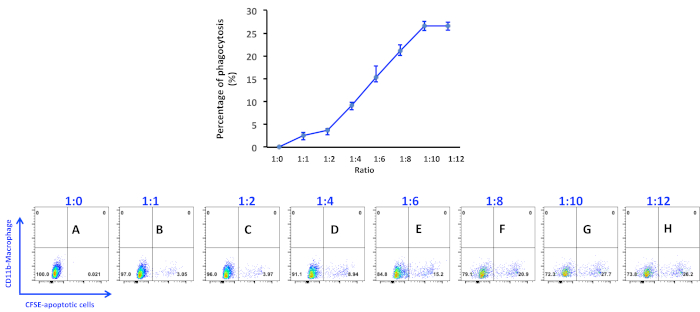
Figure 1. Percentage of phagocytosis of apoptotic thymocytes by peritoneal macrophages. Apoptotic thymocytes were induced by incubating with 1 μM of staurosporine for 4 h and added into macrophage culture at a ratio as indicated in the figure panels: (A) 1:0; (B) 1:1; (C) 1:2; (D) 1:4; (E) 1:6; (F) 1:8; (G) 1:10; (H) 1:12. Data were acquired with a flow cytometer. Percentage of phagocytic macrophages was analyzed using the software associated with the flow cytometer. Please click here to view a larger version of this figure.
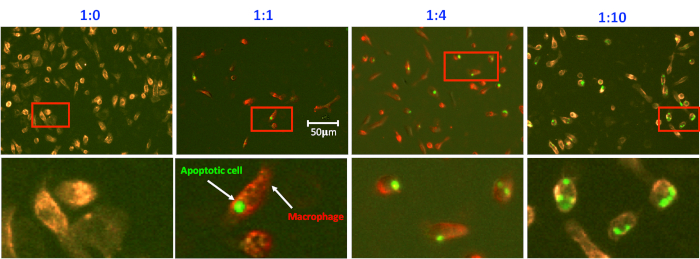
Figure 2. Microscopic analysis of efferocytosis. Efferocytosis was prepared as in Figure 1. Phagocytic macrophages were stained in situ on the plate with CD11b-PE, fixed with paraformaldehyde, and evaluated. Images were acquired using the fluorescent microscope and analyzed with the software associated with the microscope. Representative insertions were digitally enlarged and shown below in each image. Please click here to view a larger version of this figure.
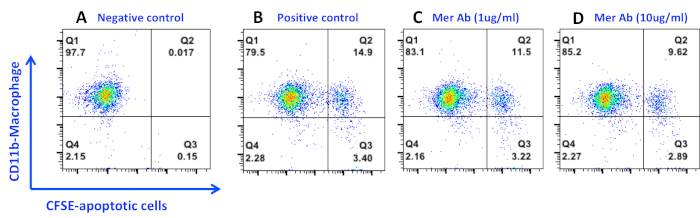
Figure 3. Inhibition of peritoneal macrophage efferocytosis by an anti-Mer antibody. Apoptotic cells were prepared and co-cultured with macrophages for 4 h as described in the protocol. Anti-Mer Ab was added into the macrophage culture immediately before the co-culture with apoptotic cells. Phagocytic macrophages were then detached and stained with anti-mouse CD11b-PE antibody on ice for 20 min. Data were acquired and analyzed as in Figure 1. Please click here to view a larger version of this figure.
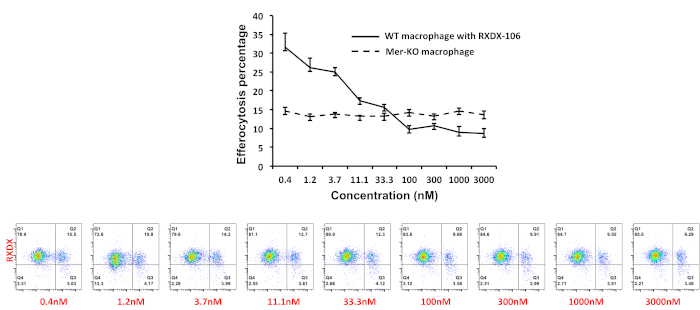
Figure 4. RXDX-106 mediated inhibition of macrophage efferocytosis. Efferocytosis was set up as described in Figure 3. RXDX-106 was added two hrs before the co-culture with apoptotic thymocytes. Mer-deficient peritoneal macrophages were prepared similarly and served as the control group. Data were acquired and the percentage of phagocytic macrophages were gated on CD11b positive cells and analyzed using the flow cytometer software. Please click here to view a larger version of this figure.
