Il recettore del fattore di crescita epidermico umano (EGFR) famiglia (ErbB) di recettori tirorosina chinasi (RTKs) comprende quattro membri – EGFR/ErbB1/HER1, ErbB2/HER2, ErbB3/HER3 e ErbB4/HER4. I recettori ErbB regolano i processi cellulari fondamentali come la crescita e la proliferazione delle cellule, la differenziazione, la migrazione e lasopravvivenza 1,2, e sono quindi potenti proto-oncogeni. L’attività aberrante dei recettori ErbB, in particolare EGFR ed ErbB2, è stata spesso associata a tumori umani che hanno reso i recettori ErbB obiettivi chiave per le terapie oncologiche2,3.
Diverse alterazioni somatiche dei geni ERBB sono state segnalate da neoplasie umane3,4,5. Gli esempi più caratterizzati includono le mutazioni ricorrenti, che attivano i punti e le brevi delezioni in-frame nel dominio della chinasi EGFR nel cancro del polmone non a piccole cellule (NSCLC). Queste mutazioni EGFR rappresentano i fattori chiave della crescita del cancro e predicono la sensibilità ai farmaciantitumoreEGFR mirati 6,7,8. Tuttavia, nella maggior parte dei tumori, le mutazioni somatiche nell’EGFR si verificano al di fuori di questi “hotspot” ricorrenti e sono distribuite nell’intera durata del recettore con 1210 residui. Infatti, la maggior parte dei residui lungo la sequenza primaria EGFR sono stati trovati per essere mutato nel cancro umano9. Tuttavia, a parte i pochi punti caldi, il significato funzionale della stragrande maggioranza delle mutazioni EGFR associate al cancro rimane sconosciuto.
La struttura monomeccatica degli ErbB è costituita da un grande dominio extracellulare amino terminale, seguito da una singola elica transmembrana che conduce al dominio della chinasi della tirosina intracellulare e alla regione della coda C-terminal che contiene siti di attracco per le proteine di segnalazione intracellulari. L’associazione Ligand innesca un drammatico cambiamento conformazionale nel dominio extracellulare, che facilita la formazione dei dimer del recettore esponendo i bracci di dimerizzazione che si incrociano simmetricamente e interagiscono con le loro superfici aromatiche/idrofobiche. Sulla formazione del recettore dei dimer i domini della tirosina chinasi entrano in contatto asimmetrico (Figura 1), con conseguente attivazione delle chinasi che fosforilate le code C-terminali dei monomeri recettori, e successivamente in attivazione della segnalazione a valle10,11.
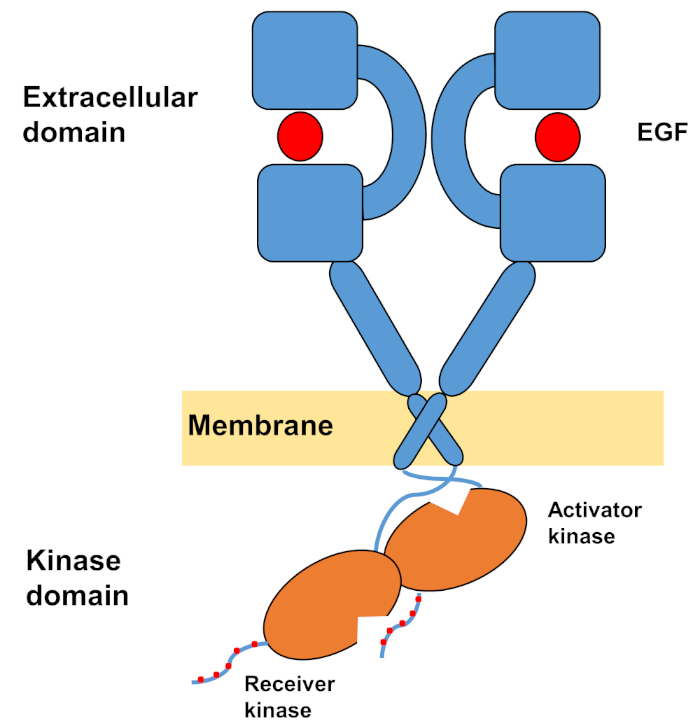
Figura 1: Struttura del dimer EGFR. EGFR si dimezza quando i domini extracellulari legano il fattore di crescita (EGF, fattore di crescita epidermico). Il dominio della chinasi del ricevitore viene quindi attivato attraverso l’interazione asimmetrica con il dominio chinasi dell’attivatore, e le code del terminale C sono autofosforilate a residui di tirosina (modificato da Tamirat et al.12). Si prega di fare clic qui per visualizzare una versione più grande di questa figura.
A causa dei riarrangiamenti strutturali dinamici che si verificano durante 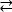 le transizioni del dimero monomero, insieme all’attivazione della chinasi associata alla formazione di un dimer asimmetrico, le mutazioni lungo l’intera lunghezza della struttura del recettore possono potenzialmente avere un effetto sulla funzione del recettore. Qui descriviamo diversi esempi dei nostri studi precedenti in cui la modellazione della mutazione e della visualizzazione erano sufficienti a spiegare le conseguenze per la funzione.
le transizioni del dimero monomero, insieme all’attivazione della chinasi associata alla formazione di un dimer asimmetrico, le mutazioni lungo l’intera lunghezza della struttura del recettore possono potenzialmente avere un effetto sulla funzione del recettore. Qui descriviamo diversi esempi dei nostri studi precedenti in cui la modellazione della mutazione e della visualizzazione erano sufficienti a spiegare le conseguenze per la funzione.
Esempio 1: Una mutazione riportata, D595V in ErbB413, ha portato ad una maggiore dimerizzazione ErbB4 e fosforilazione14. La visualizzazione della posizione della mutazione è stato un fattore critico per comprendere gli effetti funzionali osservati: D595V si è verificato al crossover simmetrico dei bracci dimerici dell’ectodomain (Figura 2A). Le braccia sono in gran parte aromatiche e idrofobiche, e la sostituzione dell’acido aspartico polare con valine dovrebbe aumentare le interazioni idrofobiche “appiccicose”, stabilizzando il dimer e quindi aumentare la lunghezza del tempo in cui avviene la fosforilazione14. All’inizio è stata una sorpresa trovare l’aspartate in ogni braccio, ma in retrospettiva si potrebbe pensare ad esso come un meccanismo di temporizzazione per l’attività, dove le catene laterali dell’acido polare riducono l’affinità e la durata del dimero intatto e quindi limitano la fosforilazione mediata dalla chinasi e la segnalazione. La sostituzione con valine rimuoverebbe quindi questa salvaguardia stabilizzando ulteriormente il dimerE ErbB4.
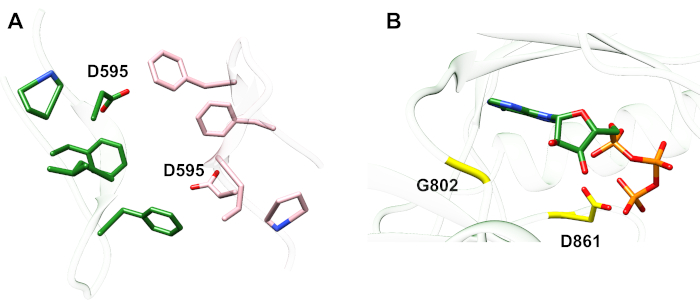
Figura 2: Posizione di una mutazione e mutazioni che attivano e di attivare la chinasi. (A) D595 (attivazione della mutazione D595V) si trova sui bracci dimerici aromatici/idrofobici del modello ectodomain ErbB4; le armi si associano al legame del fattore di crescita; (i residui vicini sono mostrati come bastoni). (B) In ErbB4, G802 (mutazione inattiva G802dup) aiuta a formare la tasca di legame intorno all’anello di adenina di ATP e D861 catalitico (mutazione D861Y inattiva) lega sia Mg2 (non mostrato) e il gruppo γ-fosfato di ATP. Si prega di fare clic qui per visualizzare una versione più grande di questa figura.
Esempio 2: Si potrebbe prevedere che le mutazioni somatiche che prendono di mira il sito di legame ATP del dominio della chinasi altererebbero o eliminerebbero l’attività ezimatica che porta a un recettore alterato o morto in chinasi incapace di segnalare. Delle nove mutazioni segnalate da pazienti con seno, gastrico, colorettale o NSCLC15, due delle nove mutazioni al momento del test avevano un’attività di fosforilazionealtamente diminuita 16: G802dup (G → GG) e D861Y. Entrambe le mutazioni somatiche inattivanti sono state trovate all’interno del sito di legame ATP della struttura di dominio della chinasi della tirosina(Figura 2B): la glicina flessibile, duplicata, altererebbe il sito dell’anello di adenina e il piccolo acido aspartico sostituito dalla tirosina ingombrante vicino ai fosfati terminali impedirebbe fisicamente la legatura di Mg2-ATP. Tuttavia, dal momento che ErbB4 può formare un eterodimero con ErbB2 – ErbB2 non lega un fattore di crescita e dipende dall’associazione con un ErbB che fa al fine di eterodimerizzare – l’ErbB2(attivo)-ErbB4(chinasi-morto) eterodimero stimolerebbe la proliferazione cellulare attraverso il percorso di segnalazione Erk/Akt, ma le cellule non si differenziano a causa della chinasi-morto ErbB4 e la mancanza di attivazione del percorso STAT516.
In studi più recenti, è diventato evidente che i movimenti dinamici degli ErbB erano rilevanti per comprendere gli effetti di alcuni mutanti sulla funzione ErbB, in particolare le mutazioni che si verificano all’interno del dominio della chinasi tirosina. Il dominio della chinasi della tirosina è costituito da un N-lobo (principalmente fogli β) e C-lobo (in gran parte alfa elicoidale), che sono separati dal sito catalitico dove si lega l’ATP. Il lobo N include l’elica e il ciclo P, mentre i loop di attivazione (A-loop) e catalitici sono presenti nel C-lobo17,18,19. Le strutture cristalline del dominio della chinasi della tirosina hanno rivelato due conformazioni inattive, la maggior parte delle strutture hanno lo stato inattivo simile a Src. Nella conformazione attiva l’aspartato catalitico dell’A-loop punta verso il sito di rilegatura ATP e l’elica di C è orientata verso la tasca di legame ATP (conformazione “C-in”), formando una forte interazione glutammato-lysina ione-coppia.
Poiché gli ErbB e il dominio della chinasi dei componenti sono entità altamente dinamiche, e soprattutto per i casi in cui gli effetti delle mutazioni sulla funzione e sull’attività biologica sono probabilmente strettamente collegati agli stati conformazionali degli ErbB, è importante valutare le mutazioni rispetto alla gamma di cambiamenti dinamici che sperimenteranno. Le strutture cristalline a raggi X degli ErbB forniscono istantanee statiche della struttura 3D, che può o non può essere rilevante per comprendere le conseguenze dinamiche di una mutazione. Al fine di sondare la gamma di cambiamenti dinamici corrispondenti al “paesaggio energetico” disponibile per una struttura tridimensionale (3D), le simulazioni di dinamiche molecolari (MD) sonoampiamente utilizzate 20. Nel caso di mutazioni che porterebbero a cambiamenti conformazionali locali all’interno del dominio della chinasi della tirosina o alla stabilizzazione di un complesso, possono essere sufficienti simulazioni dell’ordine di 100 ns. Tuttavia, le modifiche conformazionali su scala più ampia (ad esempio, le transizioni tra le conformazioni attive e inattive del dominio chinasi) richiedono un tempo di simulazione più lungo – nell’ordine dei microsecondi21.
Per quanto riguarda il protocollo descritto di seguito si considerano due mutazioni di attivazione all’interno del dominio della chinasi tirosina (Figura 3). Entrambe le mutazioni si trovano all’interno del dominio della chinasi in luoghi che sperimentano cambiamenti conformazionali locali che determinano se la chinasi è attiva o meno, e quindi le simulazioni MD sono state applicate in entrambi i casi. Nel primo caso, consideriamo le modifiche che influenzano direttamente il sito di rilegatura ATP e i macchinari catalitici del dominio della chinasi del ricevitore EGFR, esaminando specificamente le conseguenze di una mutazione di eliminazione exon 19 che è ampiamente implicata in NSCLC4,7. La mutazione ELREA750 di746, che riduce la lunghezza del loop β3-C che precede l’elica di C – l’elica che si muove verso il sito di legame/attivo sull’attivazione della chinasi e partecipa a formare l’interazione elettrostatica critica tra E762 dell’elica e K745 posizionando l’interazione lisina per l’attivazione con ATP – predispone il dominio per l’attivazione12. Nel secondo caso, consideriamo la mutazione A702V di EGFR, che si è dimostrata una nuova mutazione attivante di guadagno di funzione rivelata dalla piattaforma iScream9 e identificata in un paziente NSCLC22. Alanine-702 sul dominio chinasi ricevitore si trova sul segmento B juxtamembrane all’interfaccia del ricevitore e i domini chinasi attivatore, in cui questo asimmetrico chinasi dimer complesso e chinasi modifiche conformazionali sono necessari per l’attivazione9.
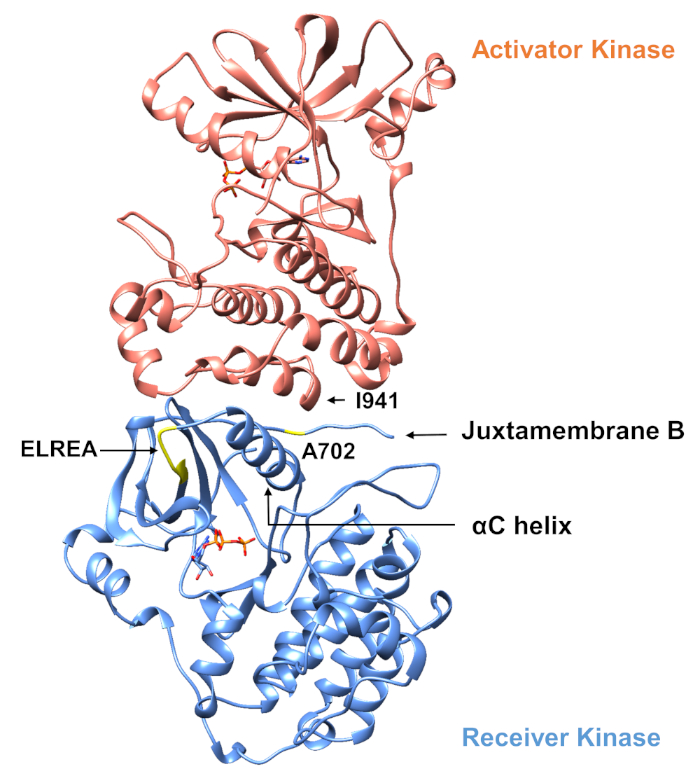
Figura 3: Il dimer di dominio della chinasi asimmetrica di EGFR. La mutazione A702V si trovava nell’interfaccia critica dei domini dell’attivatore e della chinasi del ricevitore, adiacente all’elica di C e vicino all’isolaucine 941 della chinasi dell’attivatore. I cambiamenti conformazionali indotti dalla formazione del dimer asimmetrico portano all’attivazione della chinasi. Il β3-C che contiene la sequenza ELREA precede direttamente l’elica di C; durante l’attivazione, l’elica C si sposta verso l’interno verso il sito di binding ATP. Si prega di fare clic qui per visualizzare una versione più grande di questa figura.
