Les cellules humaines sont constamment exposées à une variété d’agents d’ADN endommageant de diverses origines. Les sources exogènes consistent principalement en l’exposition aux radiations, aux produits chimiques (y compris les agents chimiothérapeutiques et certains antibiotiques) et aux virus, tandis que les principales sources endogènes comprennent des erreurs dans la réplication de l’ADN et le stress oxydatif. Les effets directs de l’exposition génotoxique peuvent aller d’une base modifiée à une rupture potentiellement mortelle à double brin d’ADN (DSB), selon le stress et la dose d’exposition. En fin de compte, les dommages non réparés ou mal réparés à l’ADN peuvent entraîner l’accumulation de mutations, les réarrangements génomiques, l’instabilité du génome et éventuellement conduire à la carcinogenèse1. Les cellules de mammifères ont développé des voies complexes pour reconnaître des types spécifiques de dommages à l’ADN2,3 et les réparer en temps opportun, synchronisés avec la progression du cycle cellulaire.
Le rayonnement ionisant (IR) endommage la double hélice de l’ADN et crée des ruptures à double brin (DSB), l’une des formes les plus nocives de dommages à l’ADN. Le complexe MRN (MRE11, RAD50, NBS1) fonctionne comme un capteur d’ADN se termine et active la protéine kinase ataxie telangiectasia muté (ATM)4,5. Suite à l’activation initiale de l’ATM par les extrémités de l’ADN, ATM déclenche une cascade d’événements DDR sur le site de la pause, initiant avec un événement clé, la phosphorylation de la variante histone H2AX6. La phosphorylation H2AX sur les résidus S139 l’active en γH2AX, couvrant les régions jusqu’à des mégabases autour de la lésion de l’ADN6,7,8,9. Cet événement augmente l’accessibilité de l’ADN, conduisant au recrutement et à l’accumulation d’autres protéines de réparation d’ADN7. Étant donné que γH2AX est abondamment et spécifiquement induit par les DSB environnants, il peut être facilement visualisé à l’aide d’anticorps spécifiques, et est couramment utilisé comme marqueur de substitution pour les DSB dans le domaine de la réparation de l’ADN. Une fois la rupture signalée, les cellules activent leurs voies de réparation de l’ADN et traitent les dommages causés par l’ADN. La protéine MDC1 (mediator de la protéine de point de contrôle des dommages de l’ADN 1) lie directement γH2AX10, interagit avec atm11 et aussi avec NBS112,13. Il contribue à accroître la concentration du complexe MRN à l’ORD et à lancer une boucle de rétroaction positive aux guichets automatiques. γH2AX est rapidement enlevé une fois la rupture réparée, ce qui permet la surveillance du dégagement d’ORD. Suivie d’une microscopie, la diminution du γH2AX au fil du temps fournit une mesure indirecte des ruptures résiduelles et de l’efficacité de la réparation de l’ADN.
Les cellules eucaryotes peuvent réparer les DSB par plusieurs voies, les deux principales étant l’assemblage final non homologue (NHEJ) et la recombinaison homologue (HR) (Figure 1). NHEJ ligates essentiellement ADN double brin se termine sans l’utilisation de l’homologie étendue et fonctionne tout au long du cycle cellulaire14,15. Hr devient prédominant pendant les phases S et G2, et est autrement réprimé, car il nécessite une sœur chromatid comme un modèle homologue pour la réparation14,16. Le choix de la voie entre le NHEJ et le HR dépend non seulement de la proximité physique du chromatid sœur, mais aussi de l’extension de la résection d’extrémité de l’ADN17, qui inhibe le NHEJ.
La réparation d’ADN homologie-dépendante initie par la dégradation nucléolytique du brin de 5′ des extrémités de rupture pour produire 3′ queues d’ADN à un brin (ssDNA), un processus appelé résection de 5′-3′. Le complexe MRN initie la résection finale de l’ADN et la résection est traitée en combinaison avec BLM/EXO1 (protéine du syndrome de Bloom/exonucléase 1) ou BLM/DNA2 (réplication de l’ADN hélicase/nucléase dépendante de l’ATP)18,19,20,21,22. La résection finale de l’ADN est améliorée par le CtIP (protéine d’interaction CtBP) par son interaction directe avec le complexeMRN 23 et le recrutement de BRCA1 (protéine de susceptibilité de type 1 du cancer du sein)24,25. La protéine de réplication A (RPA) se lie rapidement à l’ADN SsDN exposée et est ensuite déplacée par la protéine recombinase RAD51 pour former un filament de nucléoprotéine qui catalyse la recherche homologue et l’invasion de brin26,27,28.
L’initiation de la résection est une étape critique pour le choix de la voie de réparation. Une fois la résection commencée, les extrémités d’ADN deviennent de pauvres substrats pour la liaison par l’hétérodimer Ku70/Ku80 (composant de la voie NHEJ) et les cellules sont engagées à HR17,29,30. L’hétérodimère Ku70/Ku80 se lie aux extrémités de l’ORD, recrutant des ADN-PKcs et p53 Binding Protein 1 (53BP1)29,30. 53BP1 agit comme un obstacle à la résection en G1, bloquant ainsi hr tout en faisant la promotion de NHEJ31,32, mais il est supprimé d’une manière BRCA1-dépendante dans la phase S, permettant par conséquent la résection de se produire33,34. Par conséquent, 53BP1 et BRCA1 jouent un rôle opposé dans la réparation de l’ORD, avec 53BP1 étant un facilitateur NHEJ tandis que BRCA1 agit permettant des pauses pour réparer par hr.
En laboratoire, la formation d’ORD peut être induite par le rayonnement ionisant (IR). Bien que cet exemple utilise une dose élevée de 4 Gy, 1 Gy et 2 Gy également créer une quantité significative de DSBs, adapté à l’analyse de la formation de foyers par des protéines abondantes. Il est important de noter que le type et la dose de rayonnement utilisé peuvent conduire à différentes lésions dans l’ADN et dans la cellule: tandis que l’IR induit DSBs, il peut également causer des ruptures de brin unique ou la modification de la base (voir35,36 pour une référence sur le transfert d’énergie linéaire irradiation (LET) et le type de dommages à l’ADN). Pour déterminer la cinétique de la formation de foyers induits par rayonnement ionisant (IRIF) et leur dégagement, qui indiquent la réparation des dommages et l’inversion du DDRactivé 8,9,37,38, la formation de foyers peut être surveillée à différents points de temps après rayonnement ionisant. Le moment de l’activation et le dégagement de toutes les protéines majeures de dommages d’ADN est connu39, et beaucoup sont étudiés en tant que marqueurs de substitution des événements principaux. Par exemple, pRPA, qui possède une forte affinité pour l’ADN SsDN est utilisé comme un substitut de la résection de rupture, protéines MRN (MRE11, RAD50, NBS1) et exonucleases peuvent être utilisés pour évaluer l’efficacité de la résection aussi. Bien que RAD51, BRCA1, BRCA2 (protéine de susceptibilité de type 2 du cancer du sein) et PALB2 (partenaire et localisateur de BRCA2) puissent être surveillés pour évaluer l’efficacité des RH, la présence des protéines Ku ou 53BP1, sont utilisés comme marqueurs de NHEJ (Figure 1).
Comme les protéines des machines de réparation de l’ADN se recrutent les unes les autres à la rupture et s’assemblent dans des super-complexes, l’ADN-protéine et les interactions protéines-protéines peuvent être déduites en suivant leur localisation individuelle au fil du temps et en analysant la co-localisation des protéines, telle que visualisée par des signaux qui se chevauchent dans la cellule40,41,42. Dans les lignées cellulaires, l’introduction de mutations ponctuelles ou la suppression dans des gènes spécifiques de réparation de l’ADN, soit par l’édition du génome, soit par surexpression de mutants à base de plasmide, permet d’enquêter sur des résidus spécifiques et leur rôle possible dans la reconnaissance des dommages à l’ADN (p. ex., co-localisation avec γH2AX) ou d’assemblage complexe (co-localisation avec une autre ou plusieurs protéines), ainsi que leur impact sur la réparation de l’ADN. Ici, nous utilisons l’immunofluorescence indirecte comme moyen d’étudier la formation et la résolution des DSB en suivant les foyers γH2AX au fil du temps. Nous présentons également un exemple de formation de foyers et d’analyse de co-localisation par un acteur majeur de la réparation de l’ORD : p53 Binding Protein 1 (53BP1)32. Comme mentionné précédemment, 53BP1 est considéré comme central pour le choix de la voie de réparation de l’ADN. Après l’accumulation de 53BP1 et sa co-localisation avec γH2AX fournit des informations précieuses sur la phase de cycle cellulaire, l’accumulation de dommages d’ADN, et la voie utilisée pour réparer les DSB. Le but de l’immunolocalisation indirecte est d’évaluer l’efficacité de la réparation des dommages à l’ADN dans les lignées cellulaires, à la suite de l’IR comme dans cette étude, ou après l’exposition à diverses contraintes dans les cellules, de croisement d’ADN au blocage de la fourche de réplication (une liste d’agents d’endommagement de l’ADN est fournie dans le tableau 1).
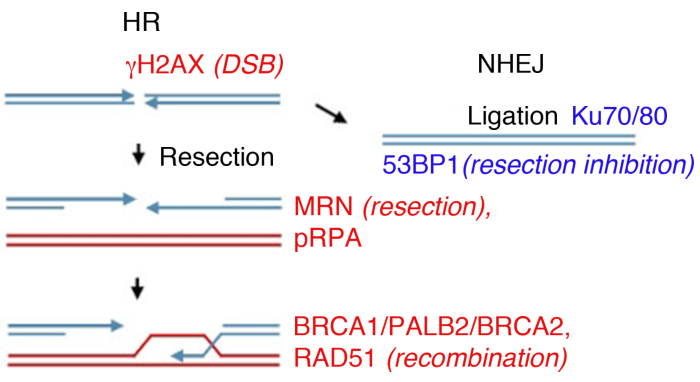
Figure 1 : Ruptures de ruptures à double brin d’ADN (DSB) voies de réparation.
La réparation d’ORD implique deux voies principales : la recombinaison homologue (HR, gauche) et l’assemblage non homologue (NHEJ, à droite). Après la pause, les protéines sont activées pour marquer la rupture (γH2AX), participer à la résection finale (MRN), enrober le ssDNA réséqué (pRPA), promouvoir la recombinaison (BRCA1, PALB2, BRCA2, RAD51) ou limiter la résection et promouvoir la SSDNA (pRPA), promouvoir la recombinaison (BRCA1, PALB2, BRCA2, RAD51) ou limiter la résection et promouvoir la resEJ (53BP1). D’autres protéines participent à la réparation des dommages, mais les protéines énumérées sont systématiquement suivies d’immunofluorescence indirecte. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
| Agent d’endommagement d’ADN | Mécanisme d’action | Dose recommandée |
| rayons γ/rayons X | Rayonnement Formation de ruptures à double brin avec certains effets cellulaires incontrôlés |
1-4 Gy |
| 36 Ions Ar | Rayonnement Formation de pauses à double brin |
270 keV/μm |
| α-particules | Rayonnement Formation de pauses à double brin |
116 keV/μm |
| Bleomycine | Inhibiteur de la synthèse de l’ADN | 0,4 à 2 μg/mL |
| Camptothécine | Inhibiteur de la topoisomérase I | 10-200 nM |
| Cisplatine | Agent alcalin (induisant des liens croisés intrastrand) |
0,25 à 2 μM |
| Doxorubicine | Agent intercalaire Inhibiteur de la topoisomerase II |
10-200 nM |
| Etoposide | Inhibiteur de la topoisomerase II | 10 μM |
| Hydroxyurea | Inhibiteur de la synthèse de l’ADN (par ribonucléotide reductase) |
10-200 μM |
| Méthane de méthyle | Agent alcalin | 0,25 à 2 mM |
| Mitomycine C | Agent alcalin | 0,25 à 2 μM |
| Lumière ultraviolette (UV) | Formation de dimers thymidine (générant une distorsion de la chaîne d’ADN) |
50-100 mJ/cm2 |
Tableau 1 : Agents génotoxiques. Exemples d’agents d’ADN nuisibles, leur mécanisme d’action et les dommages induits en fonction de la concentration de travail suggérée.
