The upright imaging method allowed us to see directly the organization of cells in the anterior follicular epithelium at stage 8. A general marker for follicle cell fate, the Eyes Absent (EYA) protein, as well as the nuclear DNA marker DAPI, showed even expression across this field of cells, and demonstrated that all cells could be seen with similar staining intensities (Figure 2B”). Proteins regulated in response to the cytokine UPD, however, showed variable patterns and expression levels. Polar cells release UPD apically prior to this stage of egg development16,17, so we expected STAT to be activated radially about the polar cells, which was sometimes the case. Often, though, we observed that downstream targets of STAT, including SLBO, had more complex expression patterns. For example, GFP reporter for SLBO expression appeared in most, but not all, cells that surrounded the polar cells (Figure 2B, and see Manning et al under review). We noticed slight differences in E-cadherin expression/localization (Figure 2B’), which may reflect changes in adhesion, but more work will be needed to quantify and interpret this result. These subtleties were not detectable through lateral three-dimensional reconstruction of staged egg chambers (Figure 2C-D). The differences in the apical-basal axis of the follicle cells can also be distinguished using the upright method. We used a reporter line for the ring canal protein Visgun (VSG), which is present in the apical region of plasma membranes18 (Figure 3A). Upright imaging confirmed VSG expression was detected in the apical portion of the follicle cells, near the narrowest part of the polar cells (Figure 3B).
We have also found that the upright imaging method works well to view the anterior epithelium in stage 9 egg chambers. At this developmental stage, the border cells coalesce together around the polar cells (Figure 4A). We could observe these compacted clusters using the end-on imaging (Figure 4B-C). As in stage 8, we found similar levels of EYA expression in all the epithelial cells (not shown), while slbo-GFP as well as activated, nuclear STAT marked the motile border cells (Figure 4B and 4C). Localization of FAS3 protein indicated the polar cell-polar cell interface (Figure 4B). In addition, in both stages 8 and 9 we could see the large nurse cells, as indicated by DAPI or phalloidin-staining of cortical actin (not shown), which suggests this method will also be useful in study of germ line cells or germ cell-follicle cell interactions.
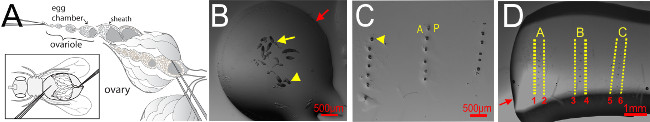
Figure 1. Upright mounting of Drosophila egg chambers. (A) Drosophila ovary indicating egg chamber, sheath, and ovariole. Depiction illustrates the position of forceps for pulling the ovariole slowly from the ovary sheath. Inset shows positioning of fly for dissection. Ventral side of female fly faces upward. A-anterior, P-posterior. (B-D) Procedure of mounting egg chambers. (B) A small drop of egg chambers in 70% glycerol-PBS, viewed under a dissection stereomicroscope. The yellow arrow indicates an ovariole chain; yellow arrowhead indicates an individual egg chamber. Red arrow indicates the edge of the Glycerol-PBS drop. (C) Columns of egg chambers arranged on thin layer of solidified glycerin jelly. Yellow arrowhead indicates an individual egg chamber. (D) A second thin layer of melted glycerin jelly covers the egg chamber columns to embed them. Yellow dotted line indicates where cuts should be made after O/N incubation at 4ᵒC. Red arrow indicates the edge of the second layer of glycerin jelly. Red numbers indicate the order of cutting the egg chamber columns (horizontal top and bottom cuts are not shown). Anterior is to the left. Please click here to view a larger version of this figure.
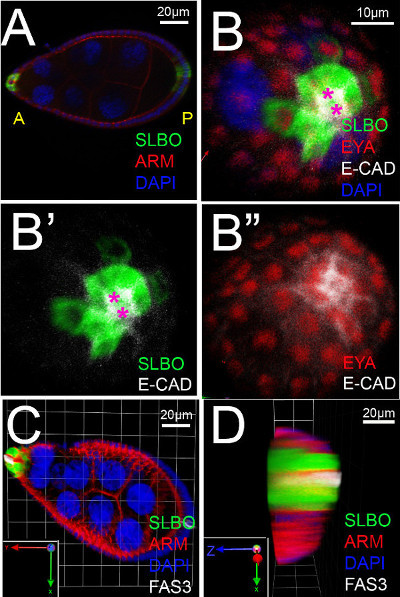
Figure 2. Imaging of the anterior epithelium of an early stage 8 egg chamber. (A) Lateral view of a stage 8 slow border cells (slbo) gene reporter egg chamber (genotype: slbo-Gal4, UAS-mCD8-GFP)19,20, stained with DAPI and primary antibodies as indicated or for slbo>GFP. Armadillo (ARM) is the fly homolog of β-catenin. Anterior (A) is to the left, posterior (P) to the right. (B-B”) End-on views of an egg chamber. Three dimensional reconstruction of Z-stack images of a stage 8 slbo-gene reporter egg chamber mounted upright, stained with primary antibodies as indicated. Magenta asterisks indicate polar cells. DAPI marks the nuclei. (B’) slbo>GFP is expressed only in the presumptive border cells (SLBO). E-cadherin (E-CAD) is an adhesion molecule localized to the membrane and enriched in border cells. (B’’) Eyes Absent (EYA) is found evenly expressed in the nuclei of all follicle cells except polar cells. (C) Standard lateral view of a slbo-reporter stage 8 egg chamber stained with DAPI (blue) and antibodies against GFP (SLBO, green), ARM (red), and FAS3 (white). Inset shows the axes: the X-axis is down (green arrow), Y-axis is towards the left (red arrow), and the Z-axis is towards the viewer (blue). (D) For comparison to the new technique, this panel shows an end on-view created by a three-dimensional reconstruction, processed with Volocity software, using a Z-stack of lateral images of the egg chamber shown in C. Inset shows the turned axes: the X-axis is down (green arrow), Z-axis is now towards the left (blue arrow), and the Y-axis is towards the viewer (red). Individual cells are blurred due to low resolution in the Z-axis; thus, the new approach shown in B represents a significant imaging improvement. Please click here to view a larger version of this figure.
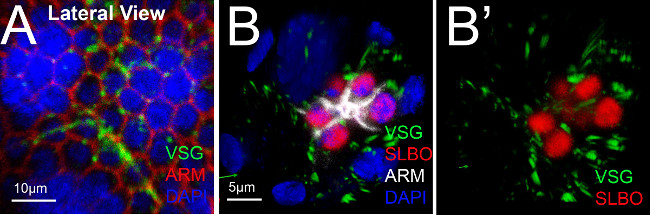
Figure 3. Comparing protein expression in lateral and anterior domains of the follicular epithelium of a stage 8 egg chamber. (A) Reconstruction of a lateral view of a stage 8 egg chamber of visgun (VSG)-GFP reporter21-23, stained for antibodies directed against GFP or ARM (white). DAPI marks the nuclei in blue, and large nurse cell nuclei are apparent. VSG localizes to the ring canals of follicle cells (green). (B-B’) End-on views of an egg chamber.Three-dimensional reconstruction of a Z-stack of images of a stage 8 VSG-GFP reporter egg chamber, stained with primary antibodies as indicated. SLBO protein (red) is detected in both polar and border cell nuclei. ARM marks follicle cell membranes and is enriched in border cells; DAPI labels nuclei. Please click here to view a larger version of this figure.
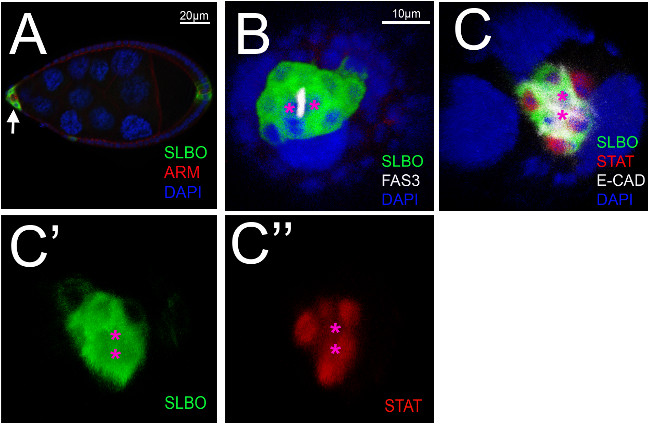
Figure 4. Imaging of early stage 9 egg chambers. (A) Lateral view of an early stage 9 slbo-reporter egg chamber. The border cells (green) have clustered within the anterior epithelium (white arrow). B-C) End-on views of the egg chambers. Three dimensional reconstruction of a Z-stack of images from an early stage 9 slbo-gene reporter egg chamber, stained using primary antibodies directed against the proteins as indicated or GFP. (B) slbo>GFP is expressed in the border cells (green), while Fasciclin 3 (FAS3, white) localizes highly to the membrane between the two polar cells; DAPI marks the nuclei (blue). Magenta asterisks indicate polar cells. (C) Both nuclear (activated) STAT and slbo>GFP expression are found only in the border cells, while E-cadherin (E-CAD) marks the membranes of follicle cells, and DAPI indicates the nuclei. Magenta asterisks indicate polar cells. Staining results are shown individually for slbo>GFP (C’) and STAT (C’’). Please click here to view a larger version of this figure.
