Highly Sensitive Assay for Measurement of Arenavirus-cell Attachment
Summary
The first step in the arenavirus life cycle is attachment of viral particles to host cells. We report a quantitative (q)RT-PCR-based assay for ultrasensitive detection and quantitation of arenavirus attachment events.
Abstract
Arenaviruses are a family of enveloped RNA viruses that cause severe human disease. The first step in the arenavirus life cycle is attachment of viral particles to host cells. While virus-cell attachment can be measured through the use of virions labeled with biotin, radioactive isotopes, or fluorescent dyes, these approaches typically require high multiplicities of infection (MOI) to enable detection of bound virus. We describe a quantitative (q)RT-PCR-based assay that measures Junin virus strain Candid 1 attachment via quantitation of virion-packaged viral genomic RNA. This assay has several advantages including its extreme sensitivity and ability to measure attachment over a large dynamic range of MOIs without the need to purify or label input virus. Importantly, this approach can be easily tailored for use with other viruses through the use of virus-specific qRT-PCR reagents. Further, this assay can be modified to permit measurement of particle endocytosis and genome uncoating. In conclusion, we describe a simple, yet robust assay for highly sensitive measurement of arenavirus-cell attachment.
Introduction
Arenaviruses are a family of enveloped, single-stranded RNA viruses that are maintained primarily in rodents in nature1-3. While these viruses typically establish an asymptomatic, persistent infection in rodent reservoirs1,2, they can cause severe disease in humans3. Important pathogenic species include the New World Junin virus (JUNV)4 and the Old World Lassa virus5, which are the etiologic agents of Argentine hemorrhagic fever in South America and Lassa fever in Africa, respectively. Lymphocytic choriomeningitis virus (LCMV), the prototypic virus of the family6, has a worldwide distribution and is responsible for a variety of disease states including aseptic meningitis in immunocompetent individuals6, severe birth defects in the developing fetus7, or high lethality in immunosuppressed individuals following solid-organ transplantation8,9. The lack of United States Food and Drug Administration (FDA)-approved vaccines or effective antivirals to combat these viruses highlights the critical need to develop novel strategies to therapeutically target these emerging/re-emerging pathogens.
The arenavirus genome consists of two single-stranded RNA segments, the large (L) (~7.2 kb) and small (S) (~3.6 kb) segments10. The S segment encodes the viral nucleoprotein (NP) and envelope glycoprotein (GP) while the L segment encodes the viral polymerase (L) and matrix protein (Z). The L and S genomic RNA segments (vRNAs) are packaged into virions10-12. The initial step of arenavirus infection is attachment of viral particles to host cells through an interaction between the viral GP and its corresponding host cell receptors which, for JUNV, includes the human transferrin receptor 1 (hTfR1)13,14. Following attachment, JUNV particles are taken into the cell via clathrin-mediated endocytosis15. Particles then traffic to the late endosome where the low pH of this compartment triggers the activation of GP16,17. Once activated, the GP-encoded fusion peptide inserts into the endosomal membrane, initiating fusion between the viral and endosomal membranes. Successful fusion results in the release of the virion-packaged vRNAs into the cytoplasm, where they serve as templates for genomic replication and transcription. When sufficient quantities of viral structural proteins and vRNAs are generated, new particles assemble and bud from the infected cell to complete the life cycle18.
To facilitate the discovery of new strategies to therapeutically target the pathogenic arenaviruses, our group has focused on the identification of host factors that are critical for virus propagation, but dispensable for the host. In this context, defining the precise stage(s) of the viral life cycle controlled by such host factors is necessary to guide the development of antiviral strategies. Herein, we describe a quantitative (q)RT-PCR-based assay that can be used to measure arenavirus-cell attachment for the purpose of identifying whether selected host proteins and/or antiviral molecules influence this initial stage of viral entry19. Alternative approaches to measure arenavirus-cell attachment have featured virions labeled with biotin20, fluorescent dyes21, or radioactive isotopes22. Drawbacks to these methods can include the requirement for large quantities of input virus (e.g. high multiplicities of infection (MOI)), the use of radioactivity, and/or additional handling steps to prepare input virus (e.g. concentration and/or labeling of virus). In particular, the use of high MOI makes it difficult to distinguish productive versus nonproductive attachment events15,23. The qRT-PCR-based assay described here can detect attachment events using as few as 20 plaque forming units (PFUs) of virus, which translates into an MOI of 0.0004 for a 48-well plate. Therefore, the strong sensitivity of the assay overcomes the requirement for high MOI as well as any virus labeling or concentration steps. Further, the assay can be i) adapted to measure virion endocytosis as well as release of virus genome into the cytoplasm following fusion and ii) tailored for use with other viruses through the development of virus-specific qRT-PCR reagents (for examples, see 24-27).
Protocol
Note: It is critical that the described protocol be carried out using strict pre-PCR technique (see 28 for an excellent overview on how to set up a pre-PCR laboratory and how to conduct experiments using good pre-PCR technique). It is imperative that all reagents and equipment are free of amplified DNA containing the qPCR target sequence and that appropriate controls are in place to detect such contamination. An overview of the protocol is shown in Figure 1.
1. Prepare Media and Seed Cells in 48-well Plates
- Prepare 500 ml of complete Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum, 1% penicillin-streptomycin, and 1% HEPES (N-2-hydroxyethylpiperazine-N-2-ethane sulfonic acid) buffer solution by adding 50 ml of heat inactivated fetal bovine serum and 5 ml each of a 100x Penicillin-Streptomycin and 100x HEPES buffer solution to a 500 ml bottle of DMEM. This reagent can be stored at 4 °C for months.
- Seed 2.5 x 104 Vero E6 cells per well in a 48-well tissue culture plate in a final volume of 500 µl of complete DMEM. Incubate the plate at 37 °C in a humidified incubator containing 5% CO2 for 10 to 18 hr.
- Seed the plate at the end of the workday. Test each virus sample in either duplicate or triplicate wells.
Note: While a 48-well-based platform for this assay is described, it should be possible to scale the assay down for use in 96- or 384-well plates.
- Seed the plate at the end of the workday. Test each virus sample in either duplicate or triplicate wells.
2. Carry Out Virus-cell Attachment
Note: The virus used in this protocol, JUNV strain Candid #1 (C#1), has been successfully used as a live attenuated vaccine for the prevention of JUNV disease29. JUNV C#1 was derived from the virulent JUNV strain XJ through serial passage both in vivo and in vitro and differs from the parental XJ strain by 12 amino acids30. Because JUNV C#1 is a biosafety level (BSL)-2-rated pathogen, the work described for this section must be carried out using appropriate BSL-2 practices31. This includes the use of personal protective equipment (e.g. eye protection, laboratory coat, double gloves), a class II biosafety cabinet, and ensuring that all personnel have received the institutional training and approvals required to safely handle this organism.
- Dilute the JUNV C#1 sample(s) to be tested to the desired PFUs or focus forming units in ice-cold complete DMEM and store on ice until ready to add to each well.
Note: As an alternative to conducting the binding studies with virus diluted in complete DMEM, use a minimal medium containing PBS with a reduced serum concentration (2%) and an appropriate buffer to establish whether serum may cause interference with virus attachment. Similar attachment protocols for alternative viruses have reported the use of RPMI 1640 medium without bicarbonate containing 0.2% bovine serum albumin, 10 mM (2-(morpholino)ethanesulfonic acid), and 10 mM HEPES, pH 6.832. This binding media may be superior to prevent pH changes that could occur when media is removed from the CO2-environment of the incubator.- Inoculate wells with PFUs ranging from 200 to 2,000 to limit nonproductive attachment events. As shown in Figure 2, this assay is capable of detecting attachment using as few as 20 PFU (or as many as 2 x 106 PFU).
- Create a master mix of virus for inoculation onto cells. Each virus sample will be inoculated onto duplicate wells in a volume of 50 µl per well. Therefore, if the addition of 2,000 PFUs of virus per well is desired, dilute 4,800 PFUs into a total volume of 120 µl of ice cold complete DMEM.
- To control for the specificity of the assay, use a matched pair of Chinese hamster ovary cell lines that either do not express TfR1 (TRVb) or express hTfR1 (TRVb-1) as JUNV attachment to these cells should either be reduced or not, respectively13,33. Alternatively, treat cells with a protease such as proteinase K to prevent arenavirus attachment34 or entry35. Soluble hTfR1 or the monocolonal antibody ch128.1, which is specific for hTfR1, could also be considered as they are known to block entry of JUNV into cells13,36. Additionally, pretreatment of cells with free mannose14 or incubation of particles with JUNV-specific neutralizing antibodies37 can inhibit virus entry.
- Please note, however, that these latter reagents and approaches, despite blocking JUNV entry, have not been confirmed to block attachment, although this is a likely explanation. The protocol described here could be used to formally determine whether these various approaches impact virion attachment.
- Remove plates from the 37 °C incubator and place either in a 4 °C refrigerator or on ice for 15 min to inhibit the ability of the plated cells to carry out endocytosis.
- Next, aspirate the complete DMEM from each well and rapidly wash each well twice using 100 µl of ice cold PBS (phosphate buffered saline) pH 7.4. Then add 50 µl of the pre-diluted virus samples (prepared in step 2.1) to the appropriate wells.
- Wrap the plate in plastic wrap and immediately place either back on ice or in a 4 °C refrigerator for 1 to 1.5 hr to allow virus attachment to occur. Keep the plate, wash buffer, and virus samples cold at 4 °C throughout this step.
- After the completion of the 4 °C incubation, aspirate the virus inoculum and wash each well 3 times with 200 µl of ice-cold PBS.
3. Viral RNA Extraction
- Purify viral RNA from JUNV C#1 particles attached to the monolayers by using the commercial kit (e.g., RNeasy Mini kit) according to manufacturer's protocol. Specifically, follow the "purification of total RNA from animal cells using spin technology protocol" on pages 23-28 in the RNeasy Mini Handbook (4th Edition, April 2006) with the modifications listed below.
- Lyse the cells as directed in step 1B by adding 350 µl of lysis buffer RLT directly to each well. Mix the buffer several times to ensure cell lysis. Note that cells easily lyse in this buffer and complete lysis can be verified by viewing wells under an inverted microscope.
- Transfer the resulting cell lysate to the kit column (step 3a) and follow the rest of the protocol as described. At this point, the virus has been neutralized by the lysis buffer so the remaining steps of the protocol can be done outside of the class II biosafety cabinet provided the kit tubes were not contaminated with infectious virus.
- Include the optional step 9 from the manufacturer's protocol, which is an additional centrifugation of the spin column to eliminate any possible carryover of Buffer RPE.
- In the final step the manufacturer's protocol (step 10), elute the purified RNA from the spin column using 30 µl of nuclease-free ddH2O. Store RNA at -80 °C at this point or use directly in the qRT-PCR assay. To prevent degradation of purified viral RNA samples by nucleases, use nuclease-free reagents and equipment and keep thawed RNA samples on ice.
Note: Buffers RLT and RW1 contain guanidine salt and should not be mixed with bleach. Buffer RW1 contains ethanol.
4. qRT-PCR Measurement of Viral Genome
- To convert JUNV C#1 S segment RNA into cDNA for subsequent qPCR, subject the viral RNA (generated in step 3.1.4) to RT in a total volume of 50 µl.
- To set up the RT reaction, first create a master mix containing each of the following reagents per reaction: 6.75 µl of nuclease-free ddH2O, 5 µl of a 2 µM stock of the RT primer S 2098- to 2075- (5'-AAGGGTTTAAAAATGGTAGCAGAC-3'), which is complementary to the NP region of the S segment genomic RNA, 5 µl of 10X PCR buffer II, 11 µl of a 25 mM MgCl2 solution, 1.25 µl (62.5 Units) of RT enzyme, 1 µl (0.4 Units) of RNase inhibitor, and 10 µl of a 500 µM stock of dNTPs.
Note: During the course of arenavirus replication, 4 viral S segment RNA species are generated: a full length genomic RNA (vRNA), a full length antigenomic RNA (vcRNA) (that is the reverse complement of the vRNA), and two subgenomic mRNAs encoding the NP and GP genes, respectively. The RT primer listed here is specific for only the S segment vRNA, but not the vcRNA, NP mRNA, or GP mRNA. - Gently mix the RT master mix and then aliquot 40 µl into individual 0.2 ml PCR tubes. Add 10 µl of viral RNA to each tube. Include i) a no template control tube (e.g. add 10 µl of nuclease-free ddH2O to 40 µl of the master mix) and ii) a no RT enzyme control (e.g. add 10 µl of a known-positive viral RNA sample to 40 µl of master mix that did not receive any RT enzyme) to screen for contamination of the RT reagents with amplified DNA containing the viral target sequence. Include RNA extracted from uninfected cells with the full RT master mix to control for assay specificity in the presence of cellular RNA.
- Additionally, include a known-positive viral RNA sample to the full master mix to verify the quality of the RT reagents. Note that the volume of the RT reaction can be reduced to 25 µl if necessary.
- To set up the RT reaction, first create a master mix containing each of the following reagents per reaction: 6.75 µl of nuclease-free ddH2O, 5 µl of a 2 µM stock of the RT primer S 2098- to 2075- (5'-AAGGGTTTAAAAATGGTAGCAGAC-3'), which is complementary to the NP region of the S segment genomic RNA, 5 µl of 10X PCR buffer II, 11 µl of a 25 mM MgCl2 solution, 1.25 µl (62.5 Units) of RT enzyme, 1 µl (0.4 Units) of RNase inhibitor, and 10 µl of a 500 µM stock of dNTPs.
- Subject the RT tubes to the following cycling conditions: 25 °C for 10 min, 48 °C for 30 min, and 95 °C for 5 min. Note that samples can be stored at -20 °C at this point or left overnight in the thermocycler by adding a 4 °C step.
- Perform qPCR in a total volume of 25 µl.
- Make a qPCR master mix by combining the following: 2.5 µl (of a 9 µM stock) of each the forward PCR primer S1937+ to 1958+ (5'-CATGGAGGTCAAACAACTTCCT-3') and reverse PCR primer S 2001- to 1982- (5'-GCCTCCAGACATGGTTGTGA-3'), 2.5 µl (of a 2 µM stock) of the qPCR probe S 1960+ to 1975+ (5'-6FAM-ATGTCATCGGATCCTT-MGBNFQ-3'), and 12.5 µl of the 2X universal qPCR master mix. Note that the forward primer reported here, which is specific for JUNV C#1, differs by 1 nt from the originally reported primer sequence38, which targets the virulent JUNV strain Romero.
- Gently mix the solution and then add 20 µl to each qPCR well. Add 5 µl of each RT reaction to the appropriate qPCR wells. Conduct qPCR in triplicate for each sample tested. Note that the PCR reaction volume can be reduced to as little as 12.5 µl if needed.
Note: The software associated with the qPCR machine will generate absolute copy numbers of JUNV C#1 S segment genomic RNA by comparing the value of each unknown sample to a series of standard dilutions of plasmid encoding the JUNV C#1 NP gene, which is the target of the qPCR primer and probe set. The qPCR assay can only provide quantitation for values that fall within the range of the standard curve. For this reason, include the following quantities of plasmid (in triplicate wells): 5, 50, 5 x 103, 5 x 105, and 5 x 107 copies per well.
- Load the qPCR plate into the thermocycler and run the following reaction conditions: 95 °C for 10 min and 40 cycles of 95 °C for 15 sec and 60 °C for 1 min.
- Collect and analyze the data to determine the quantity of viral RNA present in each sample according to manufacturer's protocol.
- To establish a standard curve using a newly purified sample of plasmid containing the qPCR target sequence, first determine the plasmid copy number in this solution.
- Calculate the molecular weight of the plasmid. If the sequence of the entire plasmid is known, calculate this using the following formula: MW = ([A * 312.2] + [G * 328.2] + [C * 288.2] + [T * 303.2] – 61.13). Remember to include the total number of a particular nucleotide found on each strand of the double stranded plasmid.
- If the size of the plasmid is known but its exact sequence is not, calculate the MW under the assumption that each dsDNA base pair has a MW of 600.
Note: The pCAGGS-JUNV C#1 NP plasmid used here has a MW of 4.2 x 106. - Once the MW of the plasmid has been determined, calculate how many copies per µl are present in the stock plasmid solution.
- First calculate the total µg of plasmid in the solution, then convert this to moles of plasmid (e.g. if 69 µg of plasmid are contained in 300 µl of water, then 6.9 x 10-5 g of plasmid * 1 mole/4.2 x 106 g of plasmid = 1.6 x 10-11 moles of plasmid), which can be converted to copy number (e.g. 1.6 x 10-11 moles of plasmid * 6.022 x 1023 molecules/mole = 9.8 x 1012 molecules (or copies)).
- Divide the total copies of plasmid in the plasmid solution by the volume of that solution to get copies per µl (e.g. 9.8 x 1012 copies/300 µl = 3.28 x 1010 copies).
- Dilute this solution appropriately so that 5 µl volumes can be loaded onto the PCR plate to deliver the range of copy numbers needed to establish the standard curve.
Representative Results
To demonstrate the sensitivity and dynamic range of the qPCR assay, known quantities of a plasmid encoding the NP gene of JUNV C#1 (range 5-5 x 107 copies) were subjected to qPCR as described in Section 4 of the protocol. The assay is sensitive and can reliably detect as few as 5 copies of the target nucleic acid (Figure 2). Further, the dynamic range of the assay is large and, as shown in Figure 2, can cover at least 7 logs of template. While not shown, we have detected as many as 5 x 109 copies of nucleic acid.
To illustrate the utility of the assay for measurement of JUNV C#1 particle attachment, various quantities of JUNV C#1 were prebound to Vero E6 cells as described in the protocol. After washing away unbound particles, cells were lysed for RNA purification and the resulting RNA samples were subjected to qRT-PCR. As shown in Figure 3, the assay can detect viral attachment events using as few as 20 or as many as 2 x 106 PFU, which translates to MOIs of 0.0004 and 40, respectively.
As shown in Figure 4, it is possible to modify the protocol to not only measure viral attachment, but also the rate of amplification of the genome contained in the incoming viral particles over time. This modified approach can be used as a surrogate to determine whether further defects in the remaining steps of viral entry (e.g. particle endocytosis and/or fusion and release of viral genome into the cytoplasm) might be occurring. Interestingly, the quantity of viral genome appears to decrease during the first 6 hr following attachment, which suggests that a portion of the incoming viral genome is degraded. This could happen to particles that fail to be taken into the cell and degrade in the extracellular space or perhaps due to degradation within the endolysosomal network. The data shown demonstrates that 12 or 18 hr post-infection would be optimal times to screen for active genome replication.
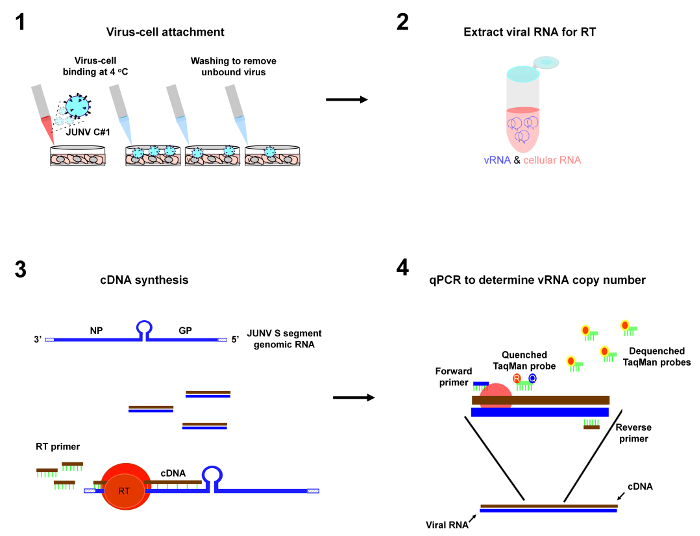
Figure 1: Depiction of protocol workflow. The primary steps of the virus-cell attachment assay are 1) incubation of virus particles with cells at 4 °C to allow virus-cell attachment to occur followed by washes to remove unbound virus, 2) purification of viral RNA from the cells, 3) reverse transcription (RT) to convert JUNV S segment genomic RNA (vRNA) into cDNA, and 4) qPCR to enumerate copies of JUNV S vRNA as a measure of virus-cell attachment. Please click here to view a larger version of this figure.
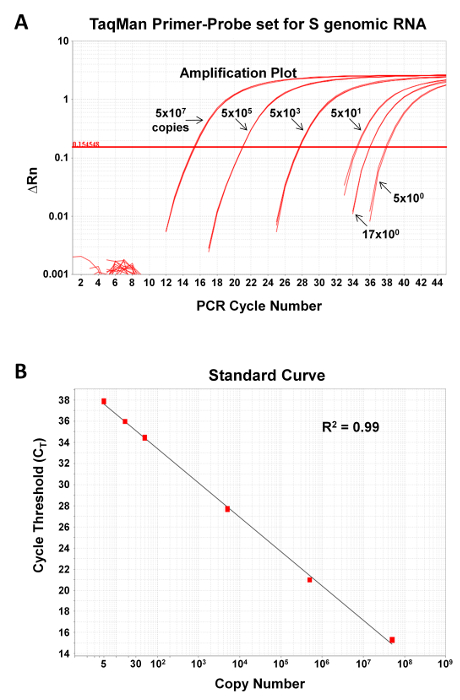
Figure 2: qPCR assay for detection of JUNV S segment genome is sensitive and can be used over a large dynamic range. The JUNV S segment qPCR primer-probe set was tested for its ability to accurately measure known quantities (5, 16.6, 50, 5 x 103, 5 x 105, or 5 x 107 copies) of a DNA standard control plasmid encoding the target sequence. Depicted are the amplification plots (A) and standard curve fit lines (B). R2, correlation coefficient of primer-probe set. Please click here to view a larger version of this figure.
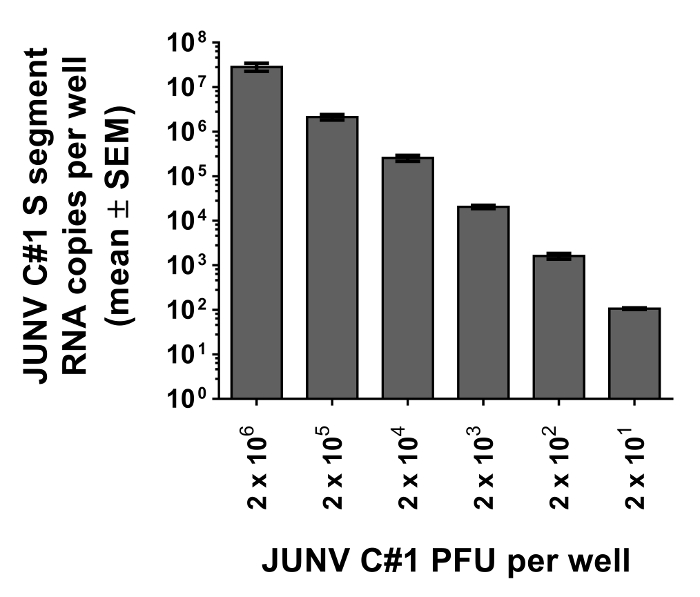
Figure 3: JUNV-cell attachment assay reliably measures particle attachment using as few as 20 PFU. Vero E6 cells were prebound with known quantities of JUNV C#1 (2 x 101, 2 x 102, 2 x 103, 2 x 104, 2 x 105, or 2 x 106 PFU) for 1.5 hr at 4 °C. Unbound particles were removed by washing, cells were lysed for RNA extraction, and the resulting RNA samples were subjected to qRT-PCR to detect JUNV C#1 S segment vRNA as a measure of particle attachment to Vero E6 cells. Data represent the mean copy number per well from duplicate wells ± SEM.
Figure 4: Kinetics of JUNV C#1 genome replication following infection. Vero E6 cells were incubated with 2 x 103 PFU of JUNV C#1 (MOI of 0.04) for 1.5 hr at 4 °C. Following several washes in ice-cold PBS, cells were either harvested immediately (0 hr time point) or warmed to 37 °C for the indicated times before collection. In each case, total RNA was extracted from cells and subjected to qRT-PCR to detect JUNV C#1 S segment vRNA. Values are listed as mean copies per well from duplicate wells ± SEM.
Discussion
The protocol we describe is straightforward and robust provided the following critical considerations are observed. First, strict pre-PCR technique must be employed (see 28 for an excellent review). It is imperative that all reagents and equipment are free of amplified DNA containing the qPCR target sequence and that appropriate controls are in place to detect such contamination. Second, for the virus-cell attachment portion of the protocol, the virus, cells, and media must be kept at 4 °C to prevent endocytosis of surface-bound virions. Third, the quantity of cells collected for RNA extraction must not exceed the RNA binding capacity of the RNA purification filter. Fourth, purified viral RNA samples must be protected from ribonuclease-mediated degradation through the use of nuclease-free reagents and equipment and by keeping them on ice when thawed. Fifth, the range of the standard curve included in the qPCR assay must be sufficiently large to capture all values observed from the unknown samples. Sixth, technical replicates should be included for both the virus-cell attachment portion of the assay as well as the qPCR of individual RNA samples. Seventh, controls (e.g. cell lines that express hTfR1 or not) can be included to control for the specificity of the assay. Finally, all personnel must have appropriate biosafety training and institutional approval to handle infectious JUNV C#1 or other pathogenic arenaviruses.
The qPCR portion of the protocol calls for a qPCR probe primarily to ensure the specificity of the product being amplified during each stage of the PCR reaction. The cost of the assay could be reduced, however, by eliminating this qPCR probe in favor of another qPCR chemistry that does not require a probe (e.g. the use of the asymmetrical dye SYBR Green I25) and/or by reducing the volume of the qPCR reaction mixture used. If a probe-independent qPCR approach is used, it would be important to verify that the reaction conditions are stringent enough to ensure specificity of the qPCR primers. It is also important to be aware that arenavirus RNA, whether extracted from cells or virions, can be nonspecifically primed to form cDNA during the RT in the absence of a virus-specific RT primer12. Therefore, copy numbers detected using a vRNA-specific RT primer do not strictly reflect just the vRNA species targeted by the RT primer. If specificity for genome or antigenome is required, we have reported a means to circumvent this nonspecific priming phenomenon through the use of biotinylated RT primers and streptavidin beads12.
An important strength of this assay is its extreme sensitivity. Other arenavirus-cell attachment assays featuring radioactively-labeled22, biotinylated20 or fluorescently-labeled21 particles have required MOIs of 0.2, 100, or 1000, respectively. While the use of radioactively-labeled particles results in good sensitivity, it necessitates the use of radioactivity, which increases the logistical complexity and safety concerns associated with this particular assay. The qPCR-based method described here requires as little as 20 PFU (MOI of ~0.0004 in a 48-well plate) and can reliably detect attachment over a large dynamic range of MOIs (e.g. MOIs from 0.0004 to at least 40) (Figure 3). The strong sensitivity of this assay makes it possible to use low MOIs for more accurate measurement of productive binding events by standard infectious virus. The high sensitivity also significantly reduces the quantity of virus needed for the assay and, in doing so, limits the additional handling steps that may impact the quality of virions being used in the assay (e.g. polyethylene glycol-based precipitation of virions, labeling virions with fluorophores or radioactive isotopes and/or sucrose-banding of virions). Additionally, the qPCR assay itself is straightforward to perform as well as highly accurate and reproducible.
The cell-attachment assay described can be modified to measure additional stages of viral entry including viral particle endocytosis as well as the final step of virus fusion, which is release of viral genome into the cytoplasm. To measure particle endocytosis, the same steps of the attachment protocol would be followed through step 2.5, which is attachment of particles to cells at 4 °C. Cells would then be warmed to allow viral particles to be endocytosed, followed by treatment with a protease to strip any externally-bound virions that were not endocytosed as described22,24. Sections 3 and 4 of this protocol would then be followed to extract viral RNA and quantitate copies of viral genomic RNA as a measure of endocytosis. To measure genome uncoating following fusion of viral and endosomal membranes, cells would be prebound with virus at 4 °C, warmed (to permit endocytosis and fusion) and stripped of external particles, and then collected for analysis. In one approach, cells could be lysed in an isotonic buffer to preserve intracellular vesicles, and then split into 2 fractions: one that would be treated with a cocktail of RNases (uncoated viral RNA is susceptible to degradation) and the other that would not39. Alternatively, cells could be separated into cytosolic or endosomal fractions40. In both cases, qRT-PCR would be used to quantitate viral genome in the cytoplasm as a measure of viral genome uncoating. Last, the qRT-PCR assay can be easily modified to study virus attachment, endocytosis, or fusion for other viruses provided the RT-PCR reagents are tailored to the new virus of interest (for examples see 24-27).
Divulgations
The authors have nothing to disclose.
Acknowledgements
We thank Markus Thali, Nathan Roy, Christopher Ziegler, Emily Bruce, and Benjamin King for helpful discussions and the National Institutes of Health for support of these studies through grants T32 AI055402 (JK), R21 AI088059 (JB), and P20RR021905 (JB).
Materials
| DMEM | Life Technologies | 11965-118 | |
| Penicillin-Streptomycin | Life Technologies | 15140-163 | 100x |
| HEPES Buffer Solution | Life Technologies | 15630-130 | 100x |
| PBS pH 7.4 | Life Technologies | 10010-023 | 1x |
| Vero E6 cells | American Type Culture Collection | CRL-1586 | |
| Costar 48-well plate | Corning | 3548 | |
| RNeasy mini kit | Qiagen | 74106 | do not mix buffers RLT or RW1 with bleach |
| QIAshredder homogenizer | Qiagen | 79654 | |
| Taqman Universal PCR Master Mix | Life Technologies | 4326614 | |
| Multiscribe Reverse Transcriptase (50U/µl) | Life Technologies | 4311235 | |
| dNTP Mixture (10 mM; 2.5 mM each dNTP) | Life Technologies | N808-0260 | |
| GeneAmp 10X PCR Buffer II and 25 mM MgCl2 solution | Life Technologies | N808-0010 | |
| RNase Inhibitor (5000U/100 µl) | Life Technologies | N808-0119 | |
| RT primer 5’-AAGGGTTTAAAAATGGTAGCAGAC-3’ | Integrated DNA Technologies | 250 nmol DNA oligo | standard desalting |
| Forward qPCR primer 5’-CATGGAGGTCAAACAACTTCCT-3’ | Life Technologies | 4304971 | standard desalting |
| Reverse qPCR primer 5’-GCCTCCAGACATGGTTGTGA-3’ | Life Technologies | 4304971 | standard desalting |
| TaqMan Probe 5’-6FAM-ATGTCATCGGATCCTT-MGBNFQ-3’ | Life Technologies | 4316033 | HPLC purified |
| MicroAmp Fast Optical 96-well reaction plate (0.1 mL) | Life Technologies | 4346906 | |
| StepOnePlus Real-Time PCR System | Life Technologies | 4376598 | or other qPCR instrument |
References
- Childs, J. C., Peters, C. J., Salvato, M. S. . The Arenaviridae. , 331-373 (1993).
- Salazar-Bravo, J., Ruedas, L. A., Yates, T. L. Mammalian reservoirs of arenaviruses. Curr. Top. Microbiol. Immunol. 262, 25-63 (2002).
- Buchmeier, M. J., de la Torre, J. C., Peters, C. J., Knipe, D. M., et al. Ch. 50. Fields Virology. 2, 1791-1827 (2007).
- Parodi, A. S., et al. Sobre la etiologia del brote epidemico de Junin (Nota previa). Dia Medico. 30, (1958).
- Frame, J. D., Baldwin, J. M., Gocke, D. J., Troup, J. M. Lassa fever, a new virus disease of man from West Africa. I. Clinical description and pathological findings. Am. J. Trop. Med. Hyg. 19, 670-676 (1970).
- Buchmeier, M. J., Zajac, A. J. . Lymphocytic Choriomenigitis Virus. , (1999).
- Barton, L. L., Mets, M. B., Beauchamp, C. L. Lymphocytic choriomeningitis virus: emerging fetal teratogen. Am. J. Obstet. Gynecol. 187, 1715-1716 (2002).
- Fischer, S. A., et al. Transmission of lymphocytic choriomeningitis virus by organ transplantation. N. Engl. J. Med. 354, 2235-2249 (2006).
- Palacios, G., et al. A new arenavirus in a cluster of fatal transplant-associated diseases. N. Engl. J. Med. 358, 991-998 (2008).
- Meyer, B. J., de la Torre, J. C., Southern, P. J. Arenaviruses: genomic RNAs, transcription, and replication. Curr. Top. Microbiol. Immunol. 262, 139-157 (2002).
- Meyer, B. J., Southern, P. J. Sequence heterogeneity in the termini of lymphocytic choriomeningitis virus genomic and antigenomic RNAs. J. Virol. 68, 7659-7664 (1994).
- Haist, K., Ziegler, C., Botten, J. Strand-Specific Quantitative Reverse Transcription-Polymerase Chain Reaction Assay for Measurement of Arenavirus Genomic and Antigenomic RNAs. PLoS One. 10, e0120043 (2015).
- Radoshitzky, S. R., et al. Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature. 446, 92-96 (2007).
- Martinez, M. G., et al. Utilization of human DC-SIGN and L-SIGN for entry and infection of host cells by the New World arenavirus, Junin virus. Biochem. Biophys. Res. Commun. 441, 612-617 (2013).
- Martinez, M. G., Cordo, S. M., Candurra, N. A. Characterization of Junin arenavirus cell entry. J. Gen. Virol. 88, 1776-1784 (2007).
- Martinez, M. G., Forlenza, M. B., Candurra, N. A. Involvement of cellular proteins in Junin arenavirus entry. Biotechnol J. 4, 866-870 (2009).
- Nunberg, J. H., York, J. The Curious Case of Arenavirus Entry, and Its Inhibition. Viruses. 4, 83-101 (2012).
- Botten, J., King, B., Klaus, J., Zielger, C. . Viral Hemorrhagic Fevers. , 233-260 (2013).
- Klaus, J. P., et al. The intracellular cargo receptor ERGIC-53 is required for the production of infectious arenavirus, coronavirus, and filovirus particles. Cell Host Microbe. 14, 522-534 (2013).
- Rojek, J. M., Perez, M., Kunz, S. Cellular entry of lymphocytic choriomeningitis virus. J. Virol. 82, 1505-1517 (2008).
- Lavanya, M., Cuevas, C. D., Thomas, M., Cherry, S., Ross, S. R. siRNA screen for genes that affect Junin virus entry uncovers voltage-gated calcium channels as a therapeutic target. Sci Transl Med. 5, 204ra131 (2013).
- Ellenberg, P., Edreira, M., Scolaro, L. Resistance to superinfection of Vero cells persistently infected with Junin virus. Arch. Virol. 149, 507-522 (2004).
- Brandenburg, B., et al. Imaging poliovirus entry in live cells. PLoS Biol. 5, e183 (2007).
- Petersen, J., et al. The Major Cellular Sterol Regulatory Pathway Is Required for Andes Virus Infection. PLoS pathogens. 10, e1003911 (2014).
- Jonsson, N., Gullberg, M., Israelsson, S., Lindberg, A. M. A rapid and efficient method for studies of virus interaction at the host cell surface using enteroviruses and real-time PCR. Virol J. 6, 217 (2009).
- Jiang, J., et al. Hepatitis C virus attachment mediated by apolipoprotein E binding to cell surface heparan sulfate. J. Virol. 86, 7256-7267 (2012).
- Shannon-Lowe, C., et al. Epstein-Barr virus-induced B-cell transformation: quantitating events from virus binding to cell outgrowth. J. Gen. Virol. 86, 3009-3019 (2005).
- Mifflin, T. E. Setting up a PCR laboratory. CSH Protoc. 2007, pdb top14 (2007).
- Maiztegui, J. I., et al. Protective efficacy of a live attenuated vaccine against Argentine hemorrhagic fever. AHF Study Group. J. Infect. Dis. 177, 277-283 (1998).
- Goni, S. E., et al. Genomic features of attenuated Junin virus vaccine strain candidate. Virus Genes. 32, 37-41 (2006).
- Chosewood, L. C., Wilson, D. E. . Biosafety in microbiological and biomedical laboratories. , (2009).
- White, J., Matlin, K., Helenius, A. Cell fusion by Semliki Forest, influenza, and vesicular stomatitis viruses. J. Cell Biol. 89, 674-679 (1981).
- McGraw, T. E., Greenfield, L., Maxfield, F. R. Functional expression of the human transferrin receptor cDNA in Chinese hamster ovary cells deficient in endogenous transferrin receptor. J. Cell Biol. 105, 207-214 (1987).
- Borrow, P., Oldstone, M. B. Characterization of lymphocytic choriomeningitis virus-binding protein(s): a candidate cellular receptor for the virus. J. Virol. 66, 7270-7281 (1992).
- Rojek, J. M., Spiropoulou, C. F., Kunz, S. Characterization of the cellular receptors for the South American hemorrhagic fever viruses Junin, Guanarito, and Machupo. 346. , 476-491 (2006).
- Helguera, G., et al. An antibody recognizing the apical domain of human transferrin receptor 1 efficiently inhibits the entry of all new world hemorrhagic Fever arenaviruses. J. Virol. 86, 4024-4028 (2012).
- Sanchez, A., et al. Junin virus monoclonal antibodies: characterization and cross-reactivity with other arenaviruses. J. Gen. Virol. 70, 1125-1132 (1989).
- Trombley, A. R., et al. Comprehensive panel of real-time TaqMan polymerase chain reaction assays for detection and absolute quantification of filoviruses, arenaviruses, and New World hantaviruses. Am. J. Trop. Med. Hyg. 82, 954-960 (2010).
- Helenius, A., Marsh, M., White, J. Inhibition of Semliki forest virus penetration by lysosomotropic weak bases. J. Gen. Virol. 58 (Pt 1), 47-61 (1982).
- Nour, A. M., Li, Y., Wolenski, J., Modis, Y. Viral membrane fusion and nucleocapsid delivery into the cytoplasm are distinct events in some flaviviruses. PLoS pathogens. 9, e1003585 (2013).
