The ability of these tools to investigate PTM crosstalk by effectively detecting these 4 PTMs is partly dependent on the unique lysis buffer system. The blastR denaturing buffer effectively isolates proteins from all cellular compartments similarly to other denaturing buffers, which ensures a complete protein profile (Figure 2A). Additionally, it preserves highly labile PTM signals like SUMO 2/3, which is rapidly diminished in non-denaturing buffers like radioimmunoprecipitation assay (RIPA) (Figure 2B). Importantly, when diluted appropriately, it does not affect the integrity of the affinity reagents like other denaturing buffers (e.g., Laemmli buffer).
A significant hurdle when working with denaturing buffers is the ability to effectively remove genomic DNA contamination. The conventional methodology to reduce viscosity is to shear the DNA by using a syringe needle or sonicating the sample. Figure 3A shows genomic DNA contamination in A431 cell lysate after treatment with the BlastR filter, syringe needle, or sonication. There is nearly complete removal of genomic DNA using the BlastR filter, which is not the case using a conventional syringe needle or sonication. Genomic DNA contamination significantly affects protein migration through an SDS- acrylamide gel; however, treatment with the BlastR filter removes genomic DNA, resulting in proper protein migration (Figure 3B). Altered migration caused by genomic DNA contamination can significantly affect interpretation of western blots; for example, the smeared EGFR pattern seen in the unfiltered lysate may be inaccurately interpreted as increased expression relative to the filtered sample (Figure 3C).
By utilizing this optimized PTM enrichment and detection system, one can rapidly determine if a target protein is modified by one or more PTMs. Investigation of the PD-L1 PTM profile was recently performed using this technique, and the results showed that PD-L1 was ubiquitinated, acetylated, and tyrosine phosphorylated in response to EGF (Figure 4). Importantly, these data reported endogenous PD-L1 PTM changes, which represented a small percentage of the total PD-L1 identified.

Figure 1: Filtering genomic DNA from cell lysate with BlastR filter. (A) Image of BlastR filter. (B) Lysate is loaded into the filter that was placed in a 15 mL collection tube. (C) Plunger is placed into the syringe and lysate is passed through the filter by compression. (D) Collect lysate, including any bubbles through complete compression. (E) Filtered lysate.
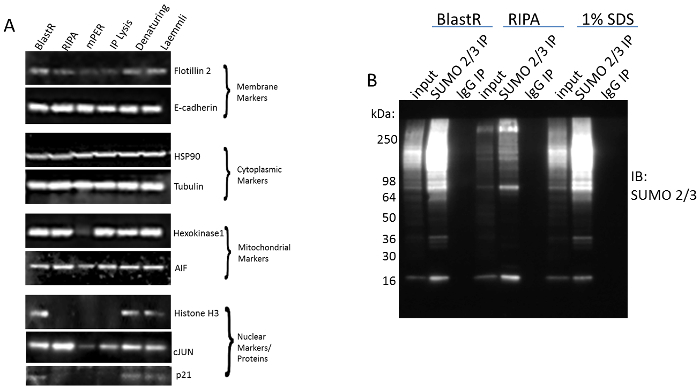
Figure 2: Comparison of BlastR lysis buffer to alternative lysis buffers. Figure 2A adapted from Horita et al. 2017. Biosci. Rep.31 (A) A431 cells were lysed with BlastR, RIPA, mPER, IP lysis, Denaturing (1% SDS), and Laemmli lysis buffers. All denaturing lysates had genomic DNA removed using BlastR filter. Isolation of proteins from the membrane, cytoplasmic, mitochondrial, and nuclear markers were determined using antibodies against the respective compartment marker proteins. (B) A431 cells were lysed with BlastR, RIPA, and 1% SDS denaturing buffer. Lysates were immunoprecipitated with SUMO 2/3 or control IgG affinity beads. Samples were separated by SDS-PAGE and analyzed by western blot using a SUMO 2/3- horseradish peroxidase (HRP) antibody. Representative blots from N≥3 independent experiments are shown. Please click here to view a larger version of this figure.
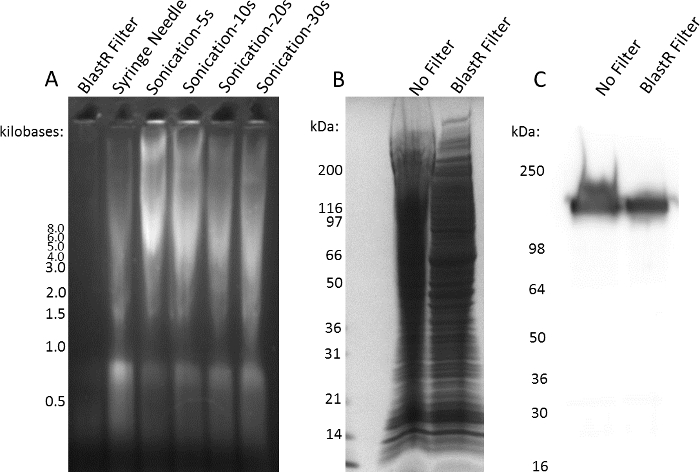
Figure 3: BlastR lysis filter is effective at removing genomic DNA. (A) A431 cells were lysed with a denaturing lysis buffer. Genomic DNA was removed or sheared with BlastR filter, syringe needle, or sonication for 5, 10, 20, or 30 seconds. 2% of the lysate was analyzed by ethidium bromide, agarose gel electrophoresis. (B) Lysate from A431 cells lysed with a denaturing buffer was either unfiltered or filter with the BlastR filter. Samples were separated with SDS-PAGE and visualized using Coomassie stain. (C) Duplicate samples from B were separated by SDS-PAGE, transferred to PVDF, and EGFR protein was examined using an EGFR antibody. Representative blots from N ≥3 independent experiments are shown. Please click here to view a larger version of this figure.
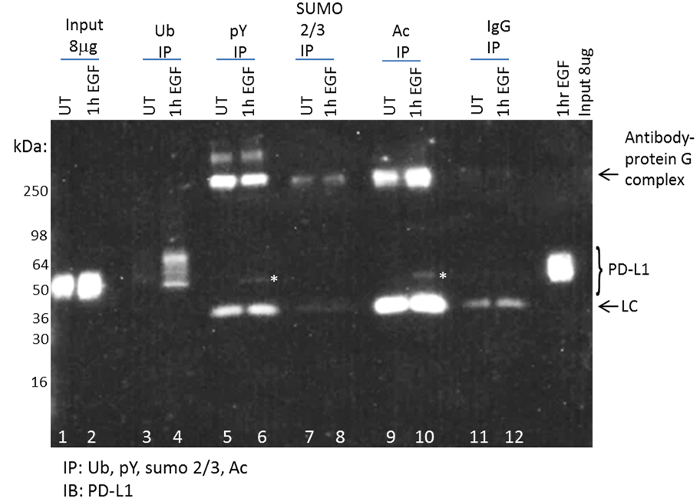
Figure 4: Detection of EGF-induced post-translational modifications for PD-L1. Figure adapted from Horita et al. 2017. Neoplasia30 (A). Serum-restricted A431 cells were either unstimulated (UT) or stimulated with EGF for one hour prior to lysis with BlastR lysis buffer. WCL was analyzed for PD-L1 levels (lanes 1,2). Ubiquitin binding beads (UBA01) were used to IP ubiquitinated proteins (lanes 3,4). Phosphotyrosine binding beads (APY03) were used to IP tyrosine-phosphorylated proteins (lanes 5,6). SUMO 2/3 binding beads (ASM24) were used to IP SUMOylated 2/3 proteins (lanes 7,8). Acetyl lysine binding beads (AAC01) were used to IP acetylated proteins (lanes 9,10). IgG binding control beads were used to IP non-specific binding proteins (lanes 11,12). Samples were separated by SDS-PAGE and analyzed by western blot using a PD-L1 antibody. A representative blot from N ≥3 independent experiments is shown. White asterisks were used to highlight PD-L1 pY and Ac protein bands. Please click here to view a larger version of this figure.
| Calculations for BlastR lysis buffer | ||||
| 1.0 mL | 2.0 mL | 5.0 mL | 10.0 mL | |
| BlastR Lysis buffer | 965 µL | 1930 µL | 4.825 mL | 9.650 mL |
| Tyrosine phosphatase Inhibitor | 5 µL | 10 µL | 25 µL | 50 µL |
| de-ubiquitinase/de-sumoylase inhibitor | 10 µL | 20 µL | 50 µL | 100 µL |
| HDAC inhibitor | 10 µL | 20 µL | 50 µL | 100 µL |
| Protease inhibitor cocktail | 10 µL | 20 µL | 50 µL | 100 µL |
| Calculations for BlastR dilution buffer | ||||
| 4.0 mL | 8.0 mL | 20.0 mL | 40.0 mL | |
| BlastR Lysis buffer | 3.86 mL | 7.72 mL | 19.3 mL | 38.6 mL |
| Tyrosine phosphatase Inhibitor | 20 µL | 40 µL | 100 µL | 200 µL |
| de-ubiquitinase/de-sumoylase inhibitor | 40 µL | 80 µL | 200 µL | 400 µL |
| HDAC inhibitor | 40 µL | 80 µL | 200 µL | 400 µL |
| Protease inhibitor cocktail | 40 µL | 80 µL | 200 µL | 400 µL |
Table 1: Lysis and Dilution Buffer Preparation Chart. Chart providing concentrations of inhibitors to add for a given lysis and dilution buffer volume when preparing buffers for cell lysis.
| Plate Protein content | Recommended BlastR Lysis Buffer volume | Recommended BlastR Dilution Buffer volume |
| < 1 mg | Combine protein from multiple plates | To make 1.5 mL final volume |
| 1-2 mg | 300 µL | To make 1.5 mL final volume |
| 2-4 mg | 600 µL | To make 3 mL final volume |
| 4-6 mg | 900 µL | To make 4.5 mL final volume |
Table 2: BlastR Lysis/Dilution Buffer Chart. Chart providing recommended lysis and dilution buffer volumes when obtaining lysate.
| Volume of cell lysate added to 1 ml of Precision Red Protein Assay reagent (µL) | Multiplier to use with sample reading OD600 |
| 10 | 10 |
| 20 | 5 |
| 30 | 3.3 |
| 40 | 2.5 |
| 50 | 2 |
Table 3: Multipliers to Convert Spectrophotometer Readings to mg/mL Lysate. Chart providing conversion numbers to aide with calculating protein concentration.
| affinity beads | volume of bead slurry/IP (mL) |
| Ubiquitination | 20 |
| Phosphotyrosine | 30 |
| SUMOylation 2/3 | 40 |
| Acetylation | 50 |
| control beads | volume of bead slurry/IP (mL) |
| Ubiquitination control beads | 20 |
| Phosphotyrsoine control beads | 30 |
| SUMOylation 2/3 control beads | 40 |
| Acetylation control beads | 50 |
Table 4: Recommended Volume of Beads/IP Chart. Chart providing recommended amounts of affinity beads to use per IP reaction.
