Vestibular and auditory sensory organs from embryonic ears, cultured in 40-Pa collagen I gels mimicking low stiffness embryonic conditions11, retain relatively normal three-dimensional structures (Figure 1) and maintain hair cells and supporting cells (Figure 2 and Figure 3). Although supporting cell density decreases by over 30% (Student's t-test: n = 4, p <0.004) and hair cell density decreases by 60% (Student's t-test: n = 5 , p <0.0001) after 3 days in utricular cultures (Figure 2), the area of the macula more than doubles over the same period of time11 (n = 3, p = 0.0002). This demonstrates that the method for three-dimensional culture described here allows for a significant increase in the number of supporting cells11, while maintaining 80% of existing hair cells in the utricle over a 3 day period. In the three-dimensional cultures established from E14.5 cochlea, Sox2-positive progenitor cells differentiate as morphologically distinguishable rows of inner and outer hair cells after 3 days in culture (Figure 3).
Gene expression can be manipulated in three-dimensional cultures of vestibular and auditory sensory organs by means of viral infection. 4hydroxytamoxifen is added to the culture medium to label supporting cells in the cochlear explants established from E15.5 embryos of Lfng-CreERT2/tdTomato mice17. Injection of adenovirus type 5 into the lumen of the culture results in infection of supporting cells at the organ's base (Figure 4A). Injection of the same virus into the lumen of utricular culture established from E17.5 embryo, results primarily in infection of supporting cells throughout the sensory epithelium (Figure 4B).
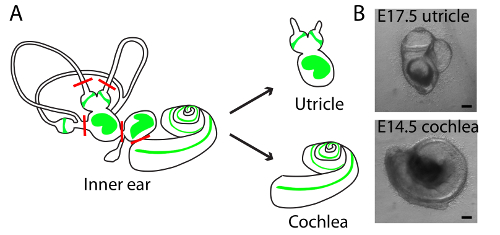
Figure 1. Schematic diagrams of dissections and light-microscopic images of representative cultures of utricle and cochlea in three-dimensional collagen I gels. (A) A schematic drawing portrays the sensory epithelia (green) of the six receptor organs of the murine inner ear. The red lines delineate the cuts introduced during the dissection of an utricle and cochlea. (B) Light microscopy images portray the E17.5 utricle (upper panel) and E14.5 cochlea (lower panel) embedded in collagen I gel and cultured for 48 h. The scale bars represent 100 µm. This figure has been modified from Gnedeva et al.11 Please click here to view a larger version of this figure.
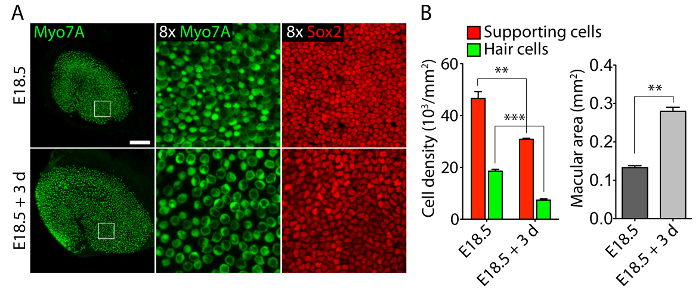
Figure 2. Hair cells and supporting cells in three-dimensional utricular cultures. (A) Confocal-microscopic images portray an E18.5 utricle prior to explantation (top panels) and after 3 days in a three-dimensional culture in 40-Pa collagen I gel (bottom panels). Hair cells are labeled for Myo7A (green) and supporting cells for Sox2 (red). The scale bar represents 50 µm. (B) Quantifications of supporting cell densities (red bars), hair cell densities (green bars), and macular areas (grey bars) in E18.5 utricles prior to explantation and after 3 days in three-dimensional organ culture are represented as means ± SEMs (p <0.001 is represented as ** and p <0.0001 as ***). This figure has been modified from Gnedeva et al.11 Please click here to view a larger version of this figure.
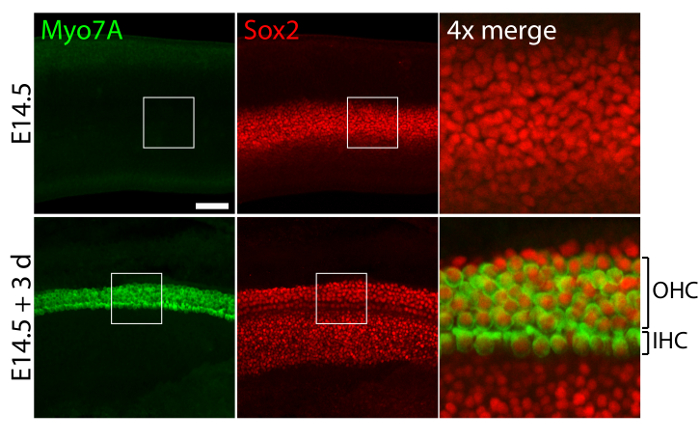
Figure 3. Hair cells and supporting cells in three-dimensional cochlear cultures. Confocal-microscopic images portray an E14.5 cochlea prior to explantation (top panels) and after 3 days in a three-dimensional culture in 40-Pa collagen I gel (bottom panels). Myo7A- and Sox2-positive inner hair cells (IHC) and outer hair cells (OHC) appear in 4 – 5 rows after 3 days in culture. Supporting cells are also labeled for Sox2 (red). The scale bar represents 25 µm. Please click here to view a larger version of this figure.
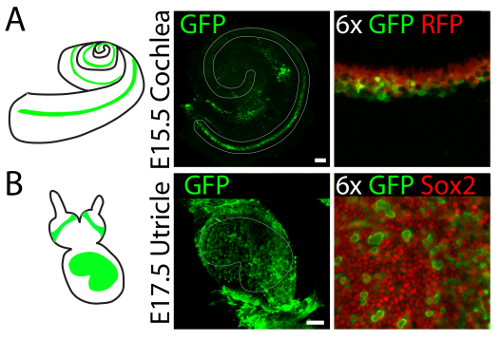
Figure 4. Representative results of viral infections in three-dimensional organ cultures of the cochlea and utricle. (A) Injection of adenovirus serotype 5 into three-dimensional cochlear cultures established from Lfng-CreERT2/tdTomato26 E15.5 embryos results in infection (GFP, green) of supporting cells (Tomato, red) at the base of the organ. The sensory epithelium is delineated in gray. The scale bar represents 100 µm. (B) An identical injection into a three-dimensional utricular culture established from an E17.5 embryo results in infection (GFP, green) of supporting cells (Sox2, red) throughout the organ. The sensory epithelium is delineated in gray. The scale bar represents 100 µm. This figure has been modified from Gnedeva et al.11 Please click here to view a larger version of this figure.
