Combined Genetic and Chemical Capsid Modifications of Adenovirus-Based Gene Transfer Vectors for Shielding and Targeting
Summary
The protocol described here enables researchers to specifically modify adenovirus capsids at selected sites by simple chemistry. Shielded adenovirus vectors particles and retargeted gene transfer vectors can be generated, and vector host interactions can be studied.
Abstract
Adenovirus vectors are potent tools for genetic vaccination and oncolytic virotherapy. However, they are prone to multiple undesired vector-host interactions, especially after in vivo delivery. It is a consensus that the limitations imposed by undesired vector-host interactions can only be overcome if defined modifications of the vector surface are performed. These modifications include shielding of the particles from unwanted interactions and targeting by the introduction of new ligands. The goal of the protocol presented here is to enable the reader to generate shielded and, if desired, retargeted human adenovirus gene transfer vectors or oncolytic viruses. The protocol will enable researchers to modify the surface of adenovirus vector capsids by specific chemical attachment of synthetic polymers, carbohydrates, lipids, or other biological or chemical moieties. It describes the cutting-edge technology of combined genetic and chemical capsid modifications, which have been shown to facilitate the understanding and overcoming of barriers for in vivo delivery of adenovirus vectors. A detailed and commented description of the crucial steps for performing specific chemical reactions with biologically active viruses or virus-derived vectors is provided. The technology described in the protocol is based on the genetic introduction of (naturally absent) cysteine residues into solvent-exposed loops of adenovirus-derived vectors. These cysteine residues provide a specific chemical reactivity that can, after production of the vectors to high titers, be exploited for highly specific and efficient covalent chemical coupling of molecules from a wide variety of substance classes to the vector particles. Importantly, this protocol can easily be adapted to perform a broad variety of different (non-thiol-based) chemical modifications of adenovirus vector capsids. Finally, it is likely that non-enveloped virus-based gene transfer vectors other than adenovirus can be modified from the basis of this protocol.
Introduction
Adenoviruses (Ad), members of the family Adenoviridae, are non-enveloped DNA viruses of which more than 70 types so far have been identified (http://hadvwg.gmu.edu). Depending on hemaglutination properties, genome structure, and sequencing results, the 70 Ad types can be divided into seven species (human adenoviruses A to G)1,2. The human Ad genome is 38 kb in size and encapsulated by an icosahedral nucleocapsid3. Due to their abundance, the capsid protein hexon, penton base, and fiber are all referred to as major capsid proteins. The most abundant and largest capsid protein hexon forms 20 capsid facets, each consisting of 12 hexon homotrimers4,5. Penton, located on each icosahedral edge (vertex), consists of pentamers of a penton base and represents the base for the vertex spike that is built of glycosylated fiber trimers5,6. The native Ad cell entry is basically composed of two major steps. First, the fiber knob binds to the primary receptor. In Ad types from species A, C, E, and F, this is the coxsackie and adenovirus receptor (CAR). This interaction brings the virion into spatial proximity of the cell surface, thereby facilitating interactions between cellular integrins and RGD-motif in the penton base and consequently inducing cellular responses. Second, changes in the cytoskeleton lead to internalization of the virion and transport to the endosome7. Upon partial disassembly in the endosome, the virion is released to the cytoplasm und ultimately travels to the nucleus for replication.
While Ad can be delivered locally (e.g., for genetic vaccination), systemic delivery through the bloodstream as required for onco-virotherapy faces several barriers. While circulating in the bloodstream, injected virions encounter the defense system of the host's immune system, leading to fast neutralization of the virus-based vectors and rendering Ad-based vectors extremely inefficient in systemic applications. Furthermore, the natural hepatotropism of Ad interferes with systemic delivery and must be resolved to redirect Ad to its new target cells.
Germline-encoded natural IgM antibodies of the innate immune system rapidly recognize and bind highly repetitive structures on the surface of the virion8,9. These immune complexes then activate the classical and non-classical pathways of the complement system, leading to fast complement-mediated neutralization of a large portion of the virions8. A second pathway resulting in major removal of Ad virions is mediated by macrophages10 and associated with acute toxic and haemodynamic side effects11,12. In the case of Ad in particular, Kupffer cells residing in the liver bind to and phagocytically take up the Ad virions via specific scavenger receptors, thereby eliminating them from the blood13,14,15. Specific scavenger receptors have also been identified on liver sinusoidal endothelial cells (LSE cells)16,and LSE cells also seem to contribute to vector elimination17, but to what extent still needs clarification. Furthermore, some Ad types and their derived vectors are efficiently sequestered by human erythrocytes18 to which they bind via CAR or the complement receptor CR119. Of note, this sequestering mechanism cannot be studied in the mouse model system as in contrast to human erythrocytes, mouse erythrocytes do not express CAR.
Specific anti-Ad antibodies generated by the adaptive immune system after exposure to Ad either due to previous infections with Ad or after the first delivery in systemic applications raise a further barrier to effective use of Ad vectors, and they should be evaded in efficient systemic delivery.
Finally, the strong hepatotropism of some Ad types (including Ad5) severely hinders application of Ad in systemic therapy. This tropism resulting in hepatocyte transduction is due to the high affinity of the Ad virion to blood coagulation factor X (FX), mediated by the interaction of FX with the Ad hexon protein20,21,22. FX bridges the virion to heparin sulfate glycans (HSPGs) on the surface of hepatocytes20,23,24,25. A crucial factor for this interaction seems to be the specific extent of N- and O-sulfation of the HSPGs in liver cells24, which is distinct from HSPGs on other cell types. In addition to this FX-mediated pathway, recent studies suggest further pathways not yet identified that result in Ad transduction of hepatocytes26,27,28.
Recently, it has been shown that FX is not only involved in hepatocyte transduction of Ad, but also by binding, the virion shields the virus particle against neutralization by the complement system26. Reduction of hepatocyte transduction by preventing FX binding, therefore, would create the unwanted side effect of increasing Ad neutralization via the innate immune system.
A profound knowledge of the complex interactions between vectors and host organisms is therefore necessary to develop more efficient vectors for systemic applications that circumvent the obstacles imposed by the host's organism.
One strategy that has been originally used for therapeutic proteins has been adapted for Ad vectors to at least partially overcome the above described barriers. Antigenicity and immunogenicity of therapeutic protein compounds could be reduced by coupling to polyethylene glycol (PEG)29,30. Hence, the covalent coupling of polymers such as PEG or poly[N-(2-hydroxypropyl)methacrylamid] (pHPMA) to the capsid surface shields the vector from unwanted vector-host interactions. Commonly, polymer coupling targets ε-amine groups from lysine side residues that are randomly distributed on the capsid surface. Vector particles in solution are, due to the hydrophilic nature of the attached polymers, surrounded by a stable water shell that reduces the risk of immune cell recognition or enzymatic degradation. Moreover, PEGylated Ad vectors were shown to evade neutralization by anti-hexon antibodies in vitro and in pre-immunized mice in vivo31. In contrast to genetic capsid modifications, chemical coupling of polymers is performed after production and purification, allowing not only for the use of conventional producer cells and production of high titer vector stocks, but also for simultaneous modification of thousands of amino acids on the capsid surface. However, amine-directed shielding occurs randomly throughout the whole capsid surface, resulting in high heterogeneities and not allowing for modification of specific capsomers. Furthermore, the large polymer moieties required for beneficial effects impair virus bioactivity32.
To overcome these limitations, Kreppel et al.33 introduced a geneti-chemical concept for vector re- and de-targeting. Cysteines were genetically introduced into the virus capsid at solvent-exposed positions like fiber HI-loop33, protein IX34, and hexon35,36. Although not naturally-occurring, cysteine-bearing Ad vectors can be produced at high titers in normal producer cells. Importantly, insertion of cysteines in certain capsomers and in different positions within a single capsomer allows for highly specific modifications of thiol group-reactive moieties. This geneti-chemical approach has been shown to overcome numerous obstacles in Ad vector design. The combination of amine-based PEGylation for detargeting and thiol-based coupling of transferrin to the fiber knob HI-loop has been proven to successfully retarget modified Ad vectors to CAR-deficient cells33. Since hexon is involved in most undesired interactions (neutralizing antibodies, blood coagulation factor FX), thiol-based modification strategies were also applied to hexon. Coupling small PEG moieties to HVR5 of hexon prevented Ad vector particles to transduce SKOV-3 cells in the presence of FX, whereas large PEG moieties increased hepatocyte transduction14,35. Ad vector particles carrying mutations in the fiber knob inhibiting CAR binding and in HVR7 inhibiting binding of FX (and bearing inserted cysteines in HVR1 for position-specific PEGylation) were shown to evade antibody- and complement-mediated neutralization, as well as scavenger receptor-mediated uptake without loss of infectivity. Interestingly, despite a lack of the natural FX shield, PEGylation again improved transduction of hepatocytes as a function of PEG size36. However, it was shown that covalent shielding does have an impact on intracellular trafficking processes. Prill et al. compared irreversible versus bioresponsive shields based on pHPMA and demonstrated that neither the mode of shielding nor co-polymer charge had an impact on cell entry but did affect particle trafficking to the nucleus. Employing a bioresponsive shield with positively charged pHPMA co-polymers allowed for particle trafficking to the nucleus, maintaining the high transduction efficiencies of Ad vectors in vitro and in vivo37.
In summary, these data indicate that, even under the assumption that all vector-host-interactions were known and considered, excessive capsid surface modifications are necessary to overcome the hurdles associated with systemic vector delivery.
Here we provide a protocol to perform site-specific chemical modifications of adenovirus vector capsids for shielding and/or retargeting of adenovirus vector particles and adenovirus-based oncolytic viruses. The concept of this technology is outlined in Figure 1. It allows the shielding of certain capsid regions from unwanted interactions by covalent attachment of synthetic polymers. At the same, it also provides a means to attach ligands and combine shielding and targeting. Using simple chemistry, experimenters will be able to covalently modify adenovirus vector surface with a wide variety of molecules including peptides/proteins, carbohydrates, lipids, and other small molecules. Furthermore, the protocol provides a general concept for the chemical modification of biologically active virus-derived vectors under maintenance of their biological integrity and activity.
Protocol
NOTE: In the following, a protocol for geneti-chemical PEGylation of an Ad vector is described to detail. To enable specific coupling of the PEG moiety, an Ad5 vector was beforehand genetically modified by introducing a cysteine residue into the hexon protein at the hypervariable loop 5 as described in a previous publication36, and a maleimide-activated PEG compound is used as coupling compound.
1. Preparation of Buffers for Vector Purification by CsCL Step Gradients
- Prepare 400 mL of Adenovirus buffer (Ad-buffer) by adding 50 mM HEPES (4.76 g/400 mL) and 150 mM NaCl (3.504 g/400 mL) to double-distilled water (ddH2O). 350 mL of this buffer is needed to prepare the following three Ad-buffer variants.
- Variant A: adjust 150 mL of Ad-buffer to pH 7.6 with NaOH.
- Variant B: add a final concentration of 10 mM TCEP [tris(2-carboxyethyl)phosphine), 0.143 g] to 50 mL and adjust it to pH 7.2 with HCl.
- Variant C: add a final concentration of 0.5 mM TCEP (0.022 g) to 150 mL and adjust it to pH 7.2 with HCl.
- Prepare the buffers in ddH2O and analytical grade chemicals. Use 100 mL each of variant A and variant C to prepare the CsCl buffers (see steps 1.2 and 1.3; 50 mL of each) necessary for the CsCl step gradients (see steps 3.2.6. and 3.2.18).
- Preparing the CsCl step gradient buffer (ρCsCl = 1.41 g/cm3) in Ad-buffer
NOTE: This buffer will be needed in two different variations:- Weigh two aliquots of exactly 27.42 g of CsCl into 50 mL tubes.
- ρCsCl = 1.41/TCEP buffer: resolve CsCl in 40 mL of variant C of Ad-buffer.
- ρCsCl = 1.41 buffer: resolve CsCl in 40 mL of variant A of Ad-buffer.
- Check, and if necessary, adjust the pH with HCl. Fill up to exactly 50 mL with the corresponding Ad-buffer variant. To achieve the best separation results, the exact CsCl concentration and pH are crucial.
- Check the density (CsCl content) by weighing 1 mL (1.41 g) of each gradient buffer.
- Preparing the CsCl step gradient buffer (ρCsCl = 1.27 g/cm3) in Ad-buffer
NOTE: This buffer is needed in two different variations:- Weigh two aliquots of exactly 18.46 g of CsCl in 50 mL tubes.
- ρCsCl = 1.27/TCEP buffer: resolve CsCl in 40 mL of variant C of Ad-buffer.
- ρCsCl = 1.27 buffer: resolve CsCl in 40 mL of variant A of Ad-buffer.
- Check, and if necessary, adjust the pH with HCl. Finally, fill up to exactly 50 mL with the corresponding Ad-buffer variant. To achieve the best separation results, the exact CsCl concentration and pH are crucial.
- Check the density (CsCl content) by weighing 1 mL (1.27 g) of each gradient buffer.
- Sterilize all buffers (Ad-buffers and CsCl step gradient buffers) by filtration (using 0.45 µm mesh pore size) into sterile bottles.
- After sterilization, to prevent oxidation of TCEP, saturate and overlay the buffers containing TCEP with argon by sparging the buffers with argon gas for about 30 s. Take care to only use sterile devices (e.g., sterile pipette tips) when applying the argon gas, and use bottles large enough to allow argon blubber.
NOTE: All buffers should be prepared fresh and protected from light and can be stored at 4 °C for several days (especially the CsCl step gradient buffers that should only be kept for a few days).
2. Coupling Moieties: Storage and Preparation
NOTE: Moeities used for coupling to cysteines need to bear thiol-reactive groups. Maleimide-activated compounds will form stable thioether bonds with the genetically introduced cysteines. Alternatively, ortho-pyridyldisulfide (OPSS)-activated compounds can be used, which form bioresponsive disulfide bridges between the vector particles and coupling moiety. Lyophilized malPEG-750 as well as most other coupling reagents are sensitive to hydrolysis and should be stored dry in the form of lyophilized powders at -80 °C.
- To prevent the build-up of condensing water upon thawing of the vials, store the vials in larger tubes (e.g., 50 mL tubes) filled with a cushion of silica gel beads. Store these tubes in an adequate larger container also filled with a silica gel bead cushion.
- Before tightly closing each tube and container, exchange air with argon gas. This can be achieved by placing the open tubes and containers into a desiccator, followed by removing and replacing the air with argon gas. Since argon gas is heavier than air, tubes and containers are filled up with argon gas and can now be placed into each other, with each lid tightly closed.
- Store containers at -80 °C.
- When thawing, place the containers onto silica beads in a desiccator and slowly warm them up to room temperature, then open the container.
- When performing a coupling reaction, freshly resolve the amount of coupling substrate needed into the appropriate solvent. Take care not to add more than 10% of coupling solution to the final coupling reaction, especially if DMF or DMSO is used as the solvent for the coupling substrate.
3. Amplification, Purification and Chemical Modification of Ad Vectors:
- Amplification of Ad vectors
NOTE: The following steps must be performed using a safety cabinet according to local biological safety rules.- Transfect a replication deficient ∆E1 Ad vector containing one (or several) genetically introduced cysteine residue(s) into HEK 293 cells as described by Kratzer and Kreppel38.
- According to the protocol38, amplify the vector by sequential reinfections up to a final preparative infection of 15-20 large (15 cm) cell culture plates (approximately 1-2 x 107 cells/plate).
NOTE: Optimally, the last reinfection should be timed so that cells show full cytopathic effect (CPE) on the morning of purification and modification performance (see Figure 2). - After the final amplification step, cells resuspended in Ad buffer + 10 mM TCEP (variant B) can be frozen at -80 °C; however, for optimal yield of vector particles, an immediate follow-up of cell lysis and vector purification and modification is recommended.
- Purification and chemical modification of Ad vectors
NOTE: Purification of vectors is performed according to the adenovirus purification protocol described in38 but under non-oxidizing conditions. Cell harvesting and lysis as well as vector purification and chemical modification can be performed within one day. Harvest and lyse the infected cells according to the protocol described previously38.- Collect cells by scraping the plates and transferring the supernatant to 200-500 mL centrifugation tubes.
- Spin the cell solution for 10 min at 400 x g.
- Resuspend the cell pellet in 4 mL of Ad-buffer + 10 mM TCEP (pH 7.2) (variant B) and transfer to a sterile 50 mL tube. If cell yield is low or only a weak CPE was induced, resuspend it in 2 mL.
- Rescue the vector by 3 cycles of freezing (in liquid nitrogen) and thawing (in a 37 °C water bath).
- Spin for 10 min at 5,000 x g at 4 °C.
- While centrifuging, prepare two equal CsCl step gradients:
- First, as the lower phase, add 3 mL of 1.41 g/cm3 ρCsCl in Ad-buffer + 0.5 mM TCEP (pH 7.2, variant C) into the ultracentrifuge tubes. Mark the level of the lower phase on the tube before loading the upper phase.
- Carefully stack the upper phase of 5 mL of 1.27 g/cm3 ρCsCl in Ad-buffer + 0.5 mM TCEP (pH 7.2, variant C) by very slowly pipetting the 5 mL volume along the walls of the tube onto the lower phase.
NOTE: Since during coupling a high concentration of TCEP could react with the maleimide residue of malPEG-750 and lead to reduced coupling efficiency, purification by CsCl step gradient needs to be already performed under a reduced TCEP concentration.
- Load the supernatant onto the CsCl gradient [either equally divide the supernatant onto both gradients or, in the case of a low vector yield (see above), load the supernatant onto only one column], and fill both gradients equally to the top with Ad buffer + 10 mM TCEP (pH 7.2, variant B).
- Adjust the weight of the opposite tubes to a 0.0 g weight difference.
- Ultracentrifuge for 2 h at 176,000 x g at 4 °C.
- With a clamp, fix the ultracentrifugation tube to a stand, leaving the area around the lower phase level mark accessible. Place the gooseneck lamp above the tube. Around the mark, at the border between the lower and upper phases, a distinct band (the vector virions) should be visible (Figure 3A-3C).
- Collect the vectors by puncturing the tube with a needle and aspiring the vector band into a syringe. Transfer collected vector virions to a 15 mL tube.
NOTE: Weaker bands above the Ad vector band contain incomplete vector particles and should be avoided for aspiration. The cell debris and green fluorescent protein (EGFP) of EGFP-expressing Ad-vectors will collect in the upper part of the ultracentrifuge tube (see Figure 3A and 3B). This and the following steps up to coupling (step 3.2.16) should be performed speedily, as the vectors are now sensible to disulfide bonding due to the low TCEP concentration. - To calculate the amount of coupling substrate needed for efficient complete binding of the substrate to all cysteine residues in the vector preparation, measure OD260:
- According to the protocol described by Kratzer and Kreppel38, vortex a mixture of 10 µL of the collected vector virions, 89 µL of Ad buffer, and 1 µL of 10% SDS. As a blank probe, vortex 99 µL of Ad-buffer and 1 µL of 10% SDS.
- To denature the virions, incubate both at 56 °C for 10 min.
- Determine OD260.
- Calculate the physical vector titer per µL using the following equation: OD260 x Factor of dilution x Empirically determined extinction coefficient of Ad (1.1 x 109 vector particles = 1 OD260 unit/µL). Therefore, a measured OD260 of 1.36 of a vector diluted 1:10, would calculate to: 1.36 x 10 x 1.1 x 109 = approximately 1.5 x 1010 vector particles/µL.
NOTE: This titer is only a rough estimation; however, it is accurate enough for the stoichiometric calculations outlined in the following.
- Depending on the protein modified by introduction of a cysteine, determine the amount of coupling substrate (in grams) for an efficient coupling reaction, according to the following general equation: Virus titer x Volume x Number of cysteines x Excess of moiety x Molecular weight of moiety / 6,022 x 1023 = grams of coupling substrate.
NOTE: In this equation: 1) virus titer is the titer/µL, determined by OD260 as described above, 2) volume accounts for the volume in µL of aspired vector used in the coupling reaction, 3) the number of cysteines is the number of proteins into which the cysteine residue was introduced per vector particle, 4) excess of moiety is the factor of moiety excess necessary for an efficient coupling reaction (50x), and 5) the molecular weight of moiety must be introduced as Da (not kDa). - Freshly resolve the amount of compound (or feasible amount) needed in the appropriate solvent.
NOTE: As the amount of coupling substrate needed is low and difficult to weigh but expensive, weigh the minimal amount of compound possible and dissolve it in an appropriate concentration for application to the coupling reaction. - Transfer the vector solution to an appropriate size tube (e.g., 2 mL tube) and add the calculated amount of coupling compound stock solution to the vector.
- Gently mix the solution by overhead rotation for 1 h at room temperature.
- Fill up the vector solution to 6 mL total volume with Ad-buffer (pH 7.6, variant A).
NOTE: This step must be performed before the solution is applied to the step gradient to ensure that the vector solution, which still contains CsCl from the first step gradient, has a density below 1.27 g/mL. - In a second CsCl banding step, purify the vector from the unreacted coupling moiety:
- Prepare two equal CsCl step gradients by performing the following steps for each: 1) Lower phase: 3 mL of 1.41 g/cm3 ρCsCl in Ad-buffer (pH 7.6, variant A), 2) mark level of lower phase on the tube before loading upper phase, and 3) upper phase: 5 mL of 1.27 g/cm3 ρCsCl in Ad-buffer (pH 7.6, variant A).
NOTE: After coupling has taken place, buffers without TCEP are used, as no free thiol groups should now be present on the vector capsids. - Load the supernatant onto the CsCl gradient and fill both gradients equally to the top with Ad-buffer (pH 7.6, variant A).
- Adjust the weights of the opposite tubes to a 0.0 g weight difference.
- Prepare two equal CsCl step gradients by performing the following steps for each: 1) Lower phase: 3 mL of 1.41 g/cm3 ρCsCl in Ad-buffer (pH 7.6, variant A), 2) mark level of lower phase on the tube before loading upper phase, and 3) upper phase: 5 mL of 1.27 g/cm3 ρCsCl in Ad-buffer (pH 7.6, variant A).
- Ultracentrifuge for 2 h at 176,000 x g at 4 °C.
- Shortly before the end of ultracentrifugation, equilibrate a PD-10 column 5 times with 5 mL of Ad-buffer (pH 7.6, variant A).
- As done in step 3.2.10, fix the ultracentrifugation tube to stand leaving area around the lower phase level mark accessible. Place the gooseneck lamp above the tube. Again, around the border between the lower and upper phases, a distinct band (the vector virions) should be visible (Figure 3C).
- Collect the vectors by puncturing the tube with a needle and aspiring the vector band into a syringe, and transfer the vector to a 15 mL tube. Take care to collect the virions of both gradient tubes together as a 2.5 mL total volume or less. If a smaller volume is collected, fill to obtain a 2.5 mL total volume with Ad buffer (variant A).
- Load the 2.5 mL onto the equilibrated PD-column. Discard the flow-through.
- Add 3 mL of Ad buffer (variant A) onto the column and collect the flow-through (containing the purified vector virions).
- Add glycerol (333 µL) to obtain a final concentration of 10% and divide into suitable aliquots (e.g., 400 µL each), and include an aliquot of 20 µL for titer determination by OD260 measurement.
- Store the vector solution at -80 °C.
- Determine the titer of vector by measuring the OD260 of 20 µL vector solution, 79 µL of Ad-buffer (variant A), and 1 µL of 10% SDS as described in step 3.2.12. As a blank, use 79 µL of Ad-buffer (variant A), 10 µL of 10% glycerol, and 1 µL of 10% SDS.
- Calculate the vector titer as: OD260 x Factor of dilution x 1.1 x 109 vector particles/µL.
As prepared in step 3.2.27, calculate: OD260 x 5 x 1.1 x 109 vector particles/µL
4. Verification of Coupling by SDS-PAGE:
NOTE: If compounds with sufficiently high molecular weights are used for coupling to Ad virions, coupling can be verified by polyacrylamide gel electrophoresis (SDS-PAGE). Successful coupling should then result in a shift of the protein band corresponding to the modified Ad virion protein, compared to the protein in the unmodified Ad virion (see Figure 4).
- According to the calculated titer of vector (step 3.2.28), separate 1-2 x 1010 vector particles by SDS-PAGE (8%).
- Develop the gel by silver-staining of the gel39 to detect even weak protein bands:
- Prepare buffers for silver staining (the amounts needed for 100 mL of buffer are indicated in brackets):
- Prepare buffer 1 by mixing 38 mL of ddH2O, 50% MeOH (50 mL of 100 MeOH), 12% AcOH (12 mL of 100% AcOH), and 0.0185% HCHO (50 µL of 37% HCHO).
- Prepare buffer 2 by adding 50% EtOH (50 mL of 100% EtOH) to 50 mL of ddH2O.
- Prepare buffer 3 by adding 0.127 g/L Na2S2O3 (12.744 mg) to 100 mL of ddH2O.
- Prepare buffer 4 by adding 2 g/L AgNO3 (0.2 g) and 0.0185% HCHO (50 µL of 37% HCHO) to 100 mL of ddH2O.
- Prepare buffer 5 by adding 60 g/L Na2CO3 (6 g), 0.0185% HCHO (50 µL of 37% HCHO), and 2.5 mg/L Na2S2O3 (0.256 mg) to ddH2O to obtain a final volume of 100 mL.
- Prepare buffer 6 by mixing 38 mL of ddH2O, 50% MeOH (50 mL of 100% MeOH), and 12% AcOH (12 mL of 100% AcOH).
- Prepare buffer 7 by mixing 60 mL of ddH2O, 30% MeOH (30 mL of 100% MeOH), and 10% glycerine (10 mL of 100% glycerine).
- Prepare buffer 8 by mixing 10% glycerine (10 mL of 100% glycerine) with 90 mL of ddH2O.
- Prepare buffers for silver staining (the amounts needed for 100 mL of buffer are indicated in brackets):
- Performing silver staining
- Fix the gel in buffer 1 for 30 min.
- Wash the gel in buffer 2 for 15 min.
- Pretreat the gel in buffer 3 for 1 min.
- Wash the gel 3 times in ddH2O for 20 s.
- Impregnate the gel in buffer 4 for 20 min.
- Wash the gel 2 times in ddH2O for 20 s.
- Develop the gel in buffer 5 until bands are visible (10 s to 10 min).
- Wash the gel 2 times in ddH2O for 2 min.
- Stop development in buffer 6 for 5 min.
- Conserve the gel in buffer 7 for 20 min.
- Store the gel in buffer 8.
NOTE: Coupling efficiency can be determined by densitometric quantification of the modified and unmodified bands of the targeted capsomere.
Representative Results
Figure 2 shows examples of the cytopathic effect (CPE) on 293 (HEK 293) cells that indicates successful vector production. Cells should show morphology (Figure 2C) 40-48 hours after inoculation with the virus vector. The right timepoint for harvesting is crucial for not losing virus particles by cell lysis and preventing oxidation of the genetically introduced thiol groups. If vector particles are released into the medium by cell lysis, the genetically introduced thiols will almost immediately become oxidized, and it will become difficult to purify and chemically modify them.
Figure 3 shows exemplary CsCl bands. The purpose of discontinuous CsCl banding before chemical modification is to remove cellular debris from the virus particles. The modification itself can then take place in CsCl. The second banding after chemical modification serves to remove an excess of (unreacted) coupling moiety. Of note, a successful modification of the virus cannot be seen in the gradient and requires further molecular analysis, ideally by SDS-PAGE.
Figure 4 shows typical examples of successful capsid modifications. Most moieties coupled to specific capsomeres will alter the running behavior of respective capsomeres in SDS-PAGE. By direct comparison with an unmodified control, the success of coupling can be verified, and densitometric analysis can help determine overall coupling efficiency (the percent of shifted and unshifted bands for the modified capsomere).
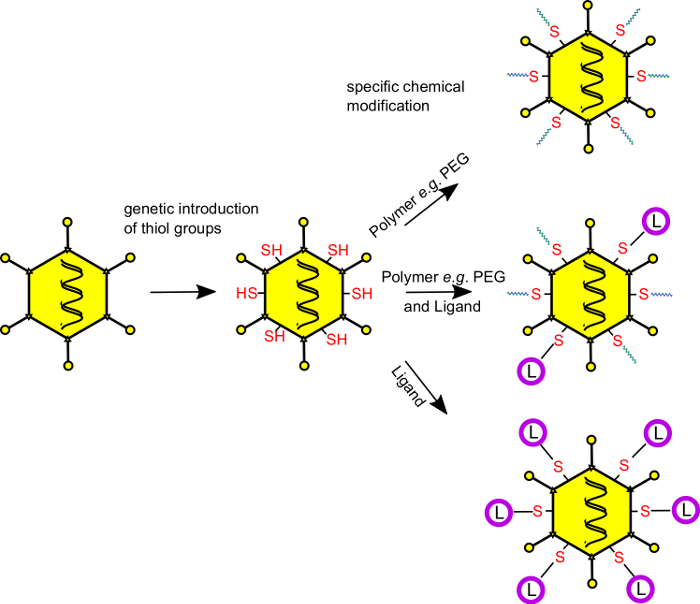
Figure 1: Combined genetic and chemical capsid modifications of adenovirus vector capsids enable covalent attachment of shielding polymers and targeting ligands. Cysteine residues are genetically introduced at selected, solvent-exposed capsid loops of adenovirus vector capsids to equip the vector particles with new chemical reactivity. The vectors can be produced using conventional producer cells and protocols. After production, the genetically introduced cysteine residues are specifically and covalently modified with thiol-reactive coupling moieties (ligands, shielding polymers, carbohydrates, small molecules, fluorescent dyes, etc.). For coupling, maleimide-activated compounds (which form stable thioether bonds with the vector particle surface) or pyridyldisulfide derivatives (which form bioresponsive disulfide bridges) can be employed. After coupling, the vectors are purified by discontinuous CsCl banding to remove unreacted excess coupling moieties. PEG, polyethylene glycol (shielding polymer); L, ligands (derived from a broad variety of substance classes); -SH, thiol functionality of the genetically introduced cysteine residues. Using this technology, a defined number of molecules can be covalently coupled to the vector capsids, and a precise selection of the coupling site is feasible. Please click here to view a larger version of this figure.
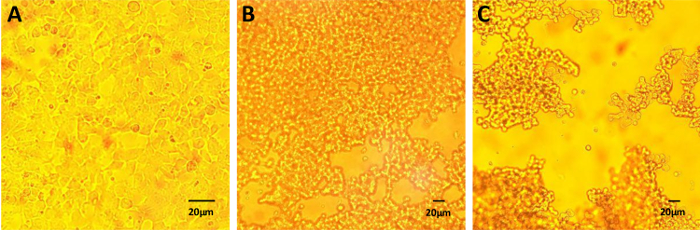
Figure 2: Cytopathic effect during vector production. Conventional producer cells (here, 293-HEK cells) can be used for production of the genetically modified vectors prior to chemical modification. The appearance of CPE indicates the best timepoint to harvest the cells. (A) Cells two hours after infection, (B) cells 24 hours after infection, and (C) cells 40 hours after infection prior to harvesting. Scale bars = 20 µm. Please click here to view a larger version of this figure.

Figure 3: Exemplary results of CsCl gradients. CsCl gradients before (A, B) and after (C) chemical modification. (A) An adenovirus vector is banded by discontinuous CsCl density gradient centrifugation. The upper phase appears green since the vector shown bears an hCMV-promoter-driven expression cassette for EGFP. The vector virion is banded as a single, white band in the lower third of the tube. The two smaller bands (above) stem from incomplete particles, which should not be collected. (B) A cysteine-bearing EGFP-expressing Ad vector before chemical modification. (C) The same vector after modification. The band in the lower third of the tube will be collected and purified by a desalting column. The pen mark in B and C labels the border of the two densities and allows for identifying the location of the vector band that will be collected. Please click here to view a larger version of this figure.

Figure 4: SiIver-stained SDS-PAGE to assess coupling efficiency. MW marker in lane 1. A vector with cysteines introduced into the capsomeres penton base, and hexon was left unmodified (lane 2) or modified with maleimide-activated PEG (polyethylene glycol) with a molecular weight of 5 kDa (lane 3). Both hexon and penton base exhibit a shift during SDS-PAGE, indicating successful modification of > 90% of the monomers per capsid. A vector with a cysteine residue in hexon was left unmodified (lane 4), modified with 5 kDa PEG (lane 5), or with 20 kDa PEG (lane 6). It should be noted that the larger shift of the hexon band is due to the higher molecular weight of the 20 kDa PEG, compared to the 5 kDa PEG (lane 5). The gel demonstrates specificity and efficiency of the coupling. Please click here to view a larger version of this figure.
Discussion
The efficiency by which the genetically introduced cysteines can be chemically modified is typically 80-99%, and certain variables influence this efficiency. First, it is paramount that the genetically introduced cysteines do not undergo premature oxidation. While being well-protected in the reducing environment of the producer cells, it is mandatory to provide a non-oxidative environment after releasing vector particles from the producer cells and during chemical modification. To this end, reducing reagents can be used at concentrations from 0.1-10 mM, and it is necessary to use reducing reagents that do not contain thiol groups, which would readily react with maleimide-activated compounds. Second, when using maleimide-activated compounds, pH during the coupling reaction should not exceed 7.35, since a pH of greater than 7.4 can decrease the specificity of the reaction. Third, in any case, amine-free buffers (e.g., HEPES, PBS) should be used for the reaction. Of note, cysteine-bearing vector particles can be purified by double CsCl banding as described, then stored in HEPES/10% glycerol prior to chemical modification. In this case, 0.1-1 mM TCEP should be used a reducing reagent, or preferably, the vectors should be stored in an argon atmosphere. Argon-filled plastic jars with lids can be used for this purpose. Additionally, modification efficiency is influenced by the hydrodynamic diameter of the compound coupled to the capsids and the site to which it is coupled. Many capsomeres that can serve as targets for the specific geneti-chemical modification are present in the capsid as trimers (fiber, hexon) or pentamers (penton base). Therefore, depending on the location of the genetically introduced cysteine, a moiety molecule coupled to one monomer might sterically hinder the coupling of another molecule to the neighboring monomer. This should be considered when selecting ideal sites for geneti-chemical capsid modifications.
To facilitate troubleshooting, a few problems may arise when following the protocol described. First, low vector yields (below 10,000 vector particles per producer cell) may result from suboptimal infection of producer cells. Ideally, 100-300 MOI should be used for the infection of producer cells. Please note that both too-high and too-low vector amounts for the inoculum generate suboptimal yields. It is ideal to passage the producer cells the day before infection. Second, smeary bands in the CsCl gradients can appear. The most frequent reason for smeary appearance of bands in the CsCl gradients is a pH of the CsCl solutions that is too acidic. The reducing reagent TCEP is very acidic, and care must be taken to adjust pH before loading the vector onto the gradient, since adenovirus vectors readily disintegrate in an acidic environment. Third, low coupling efficiencies (< 80% of coupling) are the most frequent result of impure vector preparations and/or premature oxidation of thiol groups on the vector surface. Impurities can be detected by SDS-PAGE and silver staining. In the case that significant impurities occur, vector production should be restarted. In addition, low coupling efficiencies can be caused by free non-vector thiols in the reaction. Therefore, it is advised not to use ß-mercaptoethanol or dithiothreitol as reducing reagents, since both contain free thiol groups. Of note, to select solvent-accessible sites to introduce cysteine residues that can readily be modified, crystal structures should be used; otherwise, the cysteines may not be accessible to the coupling partner. Premature oxidation of thiols on the vector surface can be avoided by the stringent use of argon and/or TCEP.
Finally, incorrect stoichiometric calculations can also lead to low coupling efficiencies. The following example is meant to illustrate the generic formula presented above and facilitate the stoichiometric calculations. In the example, one cysteine is introduced into the hexon capsomere. The titer of vector particles after the first CsCl step gradient purification is determined as 1.5 x 1010 vector particles per µL, and as coupling moiety, malPEG-750 is used. Each vector capsid contains 720 hexon proteins (240 trimers), so the titer of cysteines per µL therefore sums to 720 x 1.5 x 1010 = 1.08 x 1013 cysteines/µL. In order to modify 1.5 mL of vector particles, a total of 1.62 x 1016 cysteines must be reacted with the coupling moiety. To reach efficient coupling, a 50x molar excess of the coupling compound to the total of cysteine residues is recommended: 1.62 x 1016 x 50 = 8.1 x 1017 molecules of malPEG-750 is needed to efficiently couple to the 1.5 x 1016 cysteines of 1.5 mL of vector solution purified by the first CsCl step gradient. MalPEG-750 exhibits a molecular weight of 750 Da; hence, 6.022 x 1023 molecules of malPEG-750 weigh 750 g. Therefore, for efficient coupling of the 1.62 x 1016 cysteine residues in 1.5 mL of vector solution with a 50x excess of malPEG-750, 8.1 x 1017 molecules of malPEG-750 = (750 x 8.1 x 1017) / (6,022 x 1023) = 1 x 10-3 g = 1 mg malPEG-750 are required.
The presented method complements and exceeds the method of vector PEGylation. Conventional, amine-directed vector PEGylation does not allow for site-specific attachment of shielding or targeting moieties, whereas the geneti-chemical coupling technology presented here can be used to precisely attach moieties to adenovirus vector surfaces at defined positions and in defined copy numbers. Therefore, it is suitable for both shielding and targeting of adenovirus vector particles.
Of note, this protocol can also be used for a conventional amine-directed PEGylation of Ad vectors mentioned above. In that case, the reducing reagent TCEP can be omitted from the buffers. For stoichiometric calculations, the number of accessible amine groups per particle can be considered to be 18,000, and typical molar excesses of amine-reactive coupling moieties are 20-100 fold.
Divulgations
The authors have nothing to disclose.
Materials
| Vector purification and chemical modification | |||
| Argon gas | Air liquide | local gas dealer | |
| Liquid Nitrogen | Air liquide | local gas dealer | |
| 500 mL centrifuge tubes | Corning | 431123 | |
| Stericup Express Plus 0.22 µm | Millipore | SCGPU02RE | |
| Tris(2-carboxyethyl) phosphine (TCEP) | Sigma-Aldrich | C4706-10g | |
| 2 mL (3mL) Norm Ject (syringes) | Henke Sass Wolf | 4020.000V0 | |
| Fine-Ject needles for single use (yellow 0.9 x 40 mm) | Henke Sass Wolf | 4710009040 | |
| Caesium chloride 99.999% Ultra Quality | Roth | 8627.1 | |
| Silica gel beads | Applichem | A4569.2500 | |
| Methoxypolyethylene glycol maleimide – 750 (PEG mal-750) | Iris Biotech | store in silica gel beads at -80 °C | |
| 13.2 mL Ultra Clear Ultracentrifuge Tubes | Beckman Coulter | 344059 | only open in hood |
| PD-10 size exclusion chromatography column | GE Healthcare | 17-0851-01 | store at 4 °C |
| Hepes | AppliChem | A1069.1000 | |
| SDS Ultrapure | AppliChem | A1112,0500 | |
| Glycerol | AppliChem | A1123.1000 | |
| Name | Company | Catalog Number | Comments |
| Material for cell-culture | |||
| DPBS | PAN Biotech | P04-36500 | |
| DMEM | PAN Biotech | P04-03590 | |
| Trypsin/EDTA | PAN Biotech | P10-0231SP | |
| FBS Good | PAN Biotech | P40-37500 | |
| Penicillin/Streptomycin | PAN Biotech | P06-07100 | |
| Biosphere Filter Tips (various sizes) | Sarstedt | ||
| Serological Pipettes (various sizes) | Sarstedt | ||
| reaction tubes (various sizes) | Sarstedt | ||
| TC plates 15cm | Sarstedt | 83.3903 | |
| Name | Company | Catalog Number | Comments |
| Material for silver staining protocol | |||
| Methanol | J.T.Baker | 8045 | |
| Ethanol absolute | AppliChem | 1613,2500PE | |
| Acetic Acid | AppliChem | A0820,2500PE | |
| Formaldehyde 37% | AppliChem | A0877,0250 | |
| Ethanol absolute | AppliChem | A1613,2500PE | |
| Sodium thiosulfate | AppliChem | 1,418,791,210 | |
| Silver nitrate | AppliChem | A3944.0025 | |
| Sodium carbonate | AppliChem | A3900,0500 | |
| Name | Company | Catalog Number | Comments |
| Special Lab Equipment | |||
| Desiccator | Nalgene | 5311-0250 | |
| Megafuge 40 | Heraeus | ||
| Roter for Megafuge TX750 + Adapter andLlids for 500 mL tubes | Heraeus | ||
| Water bath | Conventional | ||
| Ultracentrifuge e.g. Optima XPN-80 | Beckman Coulter | ||
| suitable Ultrazentrifuge Rotor e.g. SW41 | Beckman Coulter | ||
| pH -Meter | Conventional | ||
| Stand with clamps | Conventional | ||
| Goose neck lamp | Conventional | ||
| Over-head rotor | Conventional | ||
| Thermal Block | Conventional | ||
| Photometer (OD 260) | Conventional |
References
- Benko, M., Harrach, B. Molecular evolution of adenoviruses. Current Topics in Microbiology and Immunology. , 3-35 (2003).
- Davison, A. J., Benko, M., Harrach, B. Genetic content and evolution of adenoviruses. Journal of Genetic Virology. 84, 2895-2908 (2003).
- Rowe, W. P., Huebner, R. J., Gilmore, L. K., Parrott, R. H., Ward, T. G. Isolation of a cytopathogenic agent from human adenoids undergoing spontaneous degeneration in tissue culture. Proceedings of the Society for Experimental Biology and Medicine. 84, 570-573 (1953).
- van Oostrum, J., Burnett, R. M. Molecular composition of the adenovirus type 2 virion. Journal of Virology. 56, 439-448 (1985).
- Stewart, P. L., Fuller, S. D., Burnett, R. M. Difference imaging of adenovirus: bridging the resolution gap between X-ray crystallography and electron microscopy. TheEMBO Journal. 12, 2589-2599 (1993).
- Stewart, P. L., Burnett, R. M., Cyrklaff, M., Fuller, S. D. Image reconstruction reveals the complex molecular organization of adenovirus. Cell. 67, 145-154 (1991).
- Meier, O., et al. Adenovirus triggers macropinocytosis and endosomal leakage together with its clathrin-mediated uptake. Journal of Cellular Biology. 158, 1119-1131 (2002).
- Xu, Z., et al. Coagulation factor X shields adenovirus type 5 from attack by natural antibodies and complement. Nature Medicine. 19, 452-457 (2013).
- Qiu, Q., et al. Impact of natural IgM concentration on gene therapy with adenovirus type 5 vectors. Journal of Virology. 89, 3412-3416 (2015).
- Alemany, R., Suzuki, K., Curiel, D. T. Blood clearance rates of adenovirus type 5 in mice. Journal of Genetic Virology. 81, 2605-2609 (2000).
- Smith, J. S., Xu, Z., Tian, J., Stevenson, S. C., Byrnes, A. P. Interaction of systemically delivered adenovirus vectors with Kupffer cells in mouse liver. Human Gene Therapy. 19, 547-554 (2008).
- Schiedner, G., et al. A hemodynamic response to intravenous adenovirus vector particles is caused by systemic Kupffer cell-mediated activation of endothelial cells. Human Gene Therapy. 14, 1631-1641 (2003).
- Xu, Z., Tian, J., Smith, J. S., Byrnes, A. P. Clearance of adenovirus by Kupffer cells is mediated by scavenger receptors, natural antibodies, and complement. Journal of Virology. 82, 11705-11713 (2008).
- Khare, R., Reddy, V. S., Nemerow, G. R., Barry, M. A. Identification of adenovirus serotype 5 hexon regions that interact with scavenger receptors. Journal of Virology. 86, 2293-2301 (2012).
- Piccolo, P., Annunziata, P., Mithbaokar, P., Brunetti-Pierri, N. SR-A and SREC-I binding peptides increase HDAd-mediated liver transduction. Gene Therapy. 21, 950-957 (2014).
- Plüddemann, A., Neyen, C., Gordon, S. Macrophage scavenger receptors and host-derived ligands. Methods (San Diego, Calif). 43, 207-217 (2007).
- Ganesan, L. P., et al. Rapid and efficient clearance of blood-borne virus by liver sinusoidal endothelium. PLoS Pathogen. 7, e1002281 (2011).
- Cichon, G., et al. Titer determination of Ad5 in blood: a cautionary note. Gene Therapy. 10, 1012-1017 (2003).
- Carlisle, R. C., et al. Human erythrocytes bind and inactivate type 5 adenovirus by presenting Coxsackie virus-adenovirus receptor and complement receptor 1. Blood. 113, 1909-1918 (2009).
- Waddington, S. N., et al. Adenovirus serotype 5 hexon mediates liver gene transfer. Cell. 132, 397-409 (2008).
- Kalyuzhniy, O., et al. Adenovirus serotype 5 hexon is critical for virus infection of hepatocytes in vivo. Proceedings of the National Academy of Sciences USA. 105, 5483-5488 (2008).
- Vigant, F., et al. Substitution of hexon hypervariable region 5 of adenovirus serotype 5 abrogates blood factor binding and limits gene transfer to liver. Molecular Therapy. The Journal of the American Society of Gene Therapy. 16, 1474-1480 (2008).
- Jonsson, M. I., et al. Coagulation factors IX and X enhance binding and infection of adenovirus types 5 and 31 in human epithelial cells. Journal of Virology. 83, 3816-3825 (2009).
- Bradshaw, A. C., et al. Requirements for receptor engagement during infection by adenovirus complexed with blood coagulation factor X. PLoS Pathogen. 6, e1001142 (2010).
- Duffy, M. R., Bradshaw, A. C., Parker, A. L., McVey, J. H., Baker, A. H. A cluster of basic amino acids in the factor X serine protease mediates surface attachment of adenovirus/FX complexes. Journal of Virology. 85, 10914-10919 (2011).
- Xu, Z., et al. Coagulation factor X shields adenovirus type 5 from attack by natural antibodies and complement. Nature Medicine. 19, 452-457 (2013).
- Prill, J. -. M., et al. Modifications of adenovirus hexon allow for either hepatocyte detargeting or targeting with potential evasion from Kupffer cells. Molecular Therapy. The Journal of the American Society of Gene Therapy. 19, 83-92 (2011).
- Zaiss, A. K., et al. Hepatocyte Heparan Sulfate Is Required for Adeno-Associated Virus 2 but Dispensable for Adenovirus 5 Liver Transduction In Vivo. Journal of Virology. 90, 412-420 (2015).
- Delgado, C., Francis, G. E., Fisher, D. The uses and properties of PEG-linked proteins. Critical Reviews in Therapeutic Drug Carrier Systems. 9, 249-304 (1992).
- Parveen, S., Sahoo, S. K. Nanomedicine: clinical applications of polyethylene glycol conjugated proteins and drugs. Clin. Pharmacokinet. 45, 965-988 (2006).
- O’Riordan, C. R., et al. PEGylation of adenovirus with retention of infectivity and protection from neutralizing antibody in vitro and in vivo. Human Gene Therapy. 10, 1349-1358 (1999).
- Subr, V., et al. Coating of adenovirus type 5 with polymers containing quaternary amines prevents binding to blood components. Journal of Controlled Release. 135, 152-158 (2009).
- Kreppel, F., Gackowski, J., Schmidt, E., Kochanek, S. Combined genetic and chemical capsid modifications enable flexible and efficient de- and retargeting of adenovirus vectors. Molecular Therapy. The Journal of the American Society of Gene Therapy. 12, 107-117 (2005).
- Corjon, S., et al. Targeting of adenovirus vectors to the LRP receptor family with the high-affinity ligand RAP via combined genetic and chemical modification of the pIX capsomere. Molecular Therapy: The Journal of the American Society of Gene Therapy. 16 (11), 1813-1824 (2008).
- Prill, J. -. M., et al. Modifications of adenovirus hexon allow for either hepatocyte detargeting or targeting with potential evasion from Kupffer cells. Molecular Therapy. The Journal of the American Society of Gene Therapy. 19, 83-92 (2011).
- Krutzke, L., et al. Substitution of blood coagulation factor X-binding to Ad5 by position-specific PEGylation: Preventing vector clearance and preserving infectivity. Journal of Controlled Release. 235, 379-392 (2016).
- Prill, J. -. M., et al. Traceless bioresponsive shielding of adenovirus hexon with HPMA copolymers maintains transduction capacity in vitro and in vivo. PloS One. 9, e82716 (2014).
- Kratzer, R. F., Kreppel, F. Production, Purification, and Titration of First-Generation Adenovirus Vectors. Methods in Molecular Biology. , 377-388 (2017).
- Blum, H., Beier, H., Gross, H. J. Improved silver staining of plant proteins, RNA and DNA in polyacrylamide gels. ELECTROPHORESIS. 8, 93-99 (1987).
