In Vivo Intracerebral Stereotaxic Injections for Optogenetic Stimulation of Long-Range Inputs in Mouse Brain Slices
Summary
This protocol describes a set of methods to identify the cell-type specific functional connectivity of long-range inputs from distant brain regions using optogenetic stimulations in ex vivo brain slices.
Abstract
Knowledge of cell-type specific synaptic connectivity is a crucial prerequisite for understanding brain-wide neuronal circuits. The functional investigation of long-range connections requires targeted recordings of single neurons combined with the specific stimulation of identified distant inputs. This is often difficult to achieve with conventional and electrical stimulation techniques, because axons from converging upstream brain areas may intermingle in the target region. The stereotaxic targeting of a specific brain region for virus-mediated expression of light-sensitive ion channels allows selective stimulation of axons originating from that region with light. Intracerebral stereotaxic injections can be used in well-delimited structures, such as the anterior thalamic nuclei, in addition to other subcortical or cortical areas throughout the brain.
Described here is a set of techniques for precise stereotaxic injection of viral vectors expressing channelrhodopsin in the mouse brain, followed by photostimulation of axon terminals in the brain slice preparation. These protocols are simple and widely applicable. In combination with whole-cell patch clamp recording from a postsynaptically connected neuron, photostimulation of axons allows the detection of functional synaptic connections, pharmacological characterization, and evaluation of their strength. In addition, biocytin filling of the recorded neuron can be used for post-hoc morphological identification of the postsynaptic neuron.
Introduction
Defining connectivity between brain regions is necessary to understand neural circuits. Classical anatomical tracing methods allow establishing interregional connectivity, and lesion studies help to understand the hierarchical organization of information flow. For example, brain circuits for spatial orientation and head direction signaling involve the directional flow of information from the thalamus to the presubiculum. This has been demonstrated by lesion studies of antero-dorsal thalamic nuclei (ADN) that degrade the head direction signal in the downstream dorsal presubiculum, as well as the parahippocampal grid cell signal1,2.
The functional connectivity between brain areas is more difficult to establish at a cellular and subcellular level. In the hippocampus, a highly organized anatomy allows to investigate pathway-specific synaptic connections using electrical simulation in the slice preparation. Stimulation electrodes placed in stratum radiatum of CA1 can be used to specifically stimulate Schaffer collateral input from CA33. Stimulating electrodes placed in stratum lacunosum moleculare of CA1 will activate the perforant path input to CA14,5. Electrical stimulation activates neurotransmitter release from axon terminals; however, it activates neurons with somata near the stimulation site as well as axons of passage. It is therefore of limited use for studying afferents from defined brain regions when fibers of different regions of origin intermingle in the target structure, as is typically the case in the neocortex.
Neurons may also be stimulated with light. Optical methods include the photoactivation of caged glutamate, which can be combined with one- or two-photon laser scanning. Multiple closely spaced sites may be stimulated sequentially, with no mechanical damage to the tissue6. This has been successfully used to map synaptic receptors as well as activate individual neurons7. While glutamate uncaging can be used for local circuit analysis, it does not allow for specific activation of long-range inputs.
A method of choice for the investigation of long-range connectivity in neuronal circuits is the use of virus-mediated channelrhodopsin expression. Using in vivo stereotaxic injections as described here, the expression of light-gated ion channels can be targeted and spatially restricted to a desired brain region. In this way, channelrhodopsins are effective for mapping excitatory or inhibitory connectivity from one region to its target. Transfected axons terminals may be stimulated with light in a brain slice preparation, and patch-clamp recordings as a read-out allow examination of the functions and strengths of specific circuit components in the brain8. The optogenetic approach combined with stereotaxic injection of a virus offers unprecedented specificity and genetic control9. Stimulating with light additionally allows for both high temporal and spatial precision10,11.
The presubiculum is a six-layered cortical structure at the transition of the hippocampus and the para-hippocampal formation12,13. It receives important synaptic input from the ADN11 but also from several other cortical and subcortical regions14. Thus, the selective stimulation of thalamic axons terminals within a presubicular slice is not possible with electrical stimulation nor glutamate uncaging. Described in this protocol are methods to determine functional connectivity between brain regions (ADN and presubiculum) using precise stereotaxic injections of viral vectors expressing light-gated channels. Also described is the photostimulation of axons terminals of projecting neurons in their target region, coupled with whole-cell patch-clamp recordings of post-synaptic neurons in the brain slice preparation.
Protocol
All procedures were performed in accordance with the European Community Council Directive (2010/63/EU) and approved by the ethics committee of Paris Descartes University. The experimenter must obtain authorization for the procedure to comply with local regulations.
1. Planning of the experiment
- Define the brain area to be targeted. Determine the stereotaxic coordinates of the injection site with the help of a mouse brain atlas15. For the right antero-dorsal thalamic nucleus (ADN), the coordinates are: -0.82 posterior, 0.75 lateral, -3.2 depth (mm) relative to bregma. Coordinates may need to be adjusted for animals of different age, sex, or strain.
- Confirm and document the exactitude of the coordinates by injecting a fluorescent tracer (150 to 300 nL) observable with an epifluorescence microscope in a pilot experiment (Figure 1A,B).
- Define the type of virus to be injected. Store the virus in 6 µL aliquots at -80 °C as recommended by the producer. Bring 1 aliquot placed on ice to the surgery room, for injection of one to six animals on a given day. Biosafety regulations for the use of AAV may depend on the country or institution, and the use of a PSM 2 hood may be required.
NOTE: Here, we use a AAV2/5 serotype expressing Chronos, a fast channelrhodospin-2 variant, fused to green fluorescent protein under the control of the Synapsin promoter: AAV5.Syn.Chronos-GFP.WPRE.bGH.
2. Stereotaxic surgery
- Install a stereotaxic frame equipped with a pump holder on a stable standard laboratory bench. Adjust stereoscope so as to clearly see the zone where the animal’s head will be placed. Use a LED light source for illumination. Rotate the stereoscope away to access the pump holder, which is not needed for the first steps of the surgery.
- Install a 10 µL Hamilton syringe equipped with a 33 G beveled metal needle in the pump holder. Test the ejection system with water.
- Anesthetize a 4- to 5-week old C57BL6 mouse with an intraperitoneal injection of a mix of ketamine hydrochloride and xylazine (100 mg/kg and 10 mg/kg, respectively). Prepare a mix of 1 mL of ketamine and 0.5 mL of xylazine in 8.5 mL of 0.9% NaCl. This will result in 10 mg/mL ketamine and 1 mg/mL xylazine in the mix. Of this mix, inject intraperitoneally 10 µL per gram of the animal’s body weight. Duration of anesthesia is about 1 h.
- Verify that the animal is well-anesthetized with a toe pinch. Then, pull out the tongue to facilitate breathing. Shave the cranial hair.
- Inject 20 µL of lidocaine hydrochloride (4 mg/mL; 2 mg/kg) under the skin of the head for local anesthesia and wait 5 min for the effect to begin. To avoid eye damage due to dryness, cover the eyes with topical ophthalmic ointment.
- To expose the skull, create a straight cut in the scalp with small surgery scissors. Place the animal in a stereotaxic frame, inserting the ear bars slightly rostral to the actual ear to rest on the bone and pull down the skin, which should create good access to the skull. Tighten into place. Install the nose piece.
- Maintain the body of the animal horizontally at the level of the head using a height-adjusted support. Place a heating pad under the mouse to keep it at physiological temperature.
- Clean the skull by applying 0.9% NaCl with a cotton swab to remove soft tissue from the bone. Use the stereoscope for the rest of the surgery.
- Adjust the skull so that the bregma-lambda axis is level, moving up or down the nose and teeth piece. This necessitates iterative measures of bregma and lamba, as both will change following adjustment of the nose level.
- Find the location of the injection site on the skull. Adjust the injection needle above the injection site according to posterior and medial coordinates and mark the skull with a disposable needle. Move the injection needle upward by 4 cm.
- Use a 0.5 mm burr with a drill to realize a 1 mm diameter craniotomy on the mark, at one-half of maximum speed. Swab eventual bleeding with a paper tissue.
- Empty the water contained in the Hamilton syringe for storage by completely ejecting it with the pump. Only the needle will still be filled with water. The needle is washed between each use with pure deionized water. Sterilization is not required.
- Take the aliquot of virus that is to be used for this day. Make sure that the viral solution is not frozen anymore but has remained cooled (close to 0 °C, on ice). Only briefly remove from the ice to obtain 700 nL with a micropipette for small volumes. Deposit the drop on a 5 cm x 5 cm piece of paraffin film. Avoid creating bubbles. Put the remaining viral solution back on ice.
NOTE: The drop volume should be greater than the desired injection volume (700 nL for 200 nL injected). This will give a safety margin in case some of the liquid is lost during the transfer and allows performing a small test ejection (step 2.16) before proceeding. - Place the paraffin film on top of the craniotomy. Plunge the needle in the drop of viral solution without changing the antero-posterior and lateral position.
- Use the "withdraw" function of the pump to fill the syringe with about 500 nL of viral solution disposed on the paraffin film. Do this under visual control (stereoscope), watch the drop disappear, and make sure not to aspirate air.
- Make sure the syringe has been filled correctly. Verify the functioning of the ejection system by driving down the plunger to test eject a small drop of liquid of 50 nL under visual control. Wipe the drop.
- Insert the needle into the brain to the chosen depth, by turning the knob controlling the dorso-ventral axis of the stereotaxic apparatus clockwise. Push the "run" button (speed 15 nL/min per volume of 150 nL injected). A small volume (50-300 nL, depending on the virus used) is slowly ejected over 10 min with an automatic pump.
- Wait 10 min after the injection to avoid leaking from the injection site. Then, slowly remove the needle over 3-5 min by turning the knob controlling the dorso-ventral axis counterclockwise.
- Rotate the vertical part of the stereotaxic frame with the syringe away from the animal. Immediately wash the needle in clean distilled water by filling-emptying it several times, in order to avoid clogging. Store the syringe filled with water.
- Remove the mouse from the stereotaxic frame. Suture the skin with 4-0 polyamide suture filament. Make three or four stiches, tied with 2-1-1 standard surgical knots.
- Place the mouse in a heated cage until it completely wakes up from anesthesia, and provide water and soaked food in a Petri dish placed on the ground. If the heat source is below the cage, use a spacer grid to avoid overheating.
NOTE: According to local guidelines, a single dose of ketoprofen (2-5 mg/kg, subcutaneously) or buprenorphine (0.05-0.1 mg/kg, subcutaneously) may be applied to prevent pain. - When the animal is fully awake, return it to its home cage and monitor its well-being, particularly on the day following injection. Check for signs of pain. If any behavioral modification is observed, the animal is weighed to monitor its body weight.
- Depending on the virus used, the time for full expression may vary. Here, we allow 3 weeks for expression of AAV5.Syn.Chronos-GFP.
3. Solutions for acute slice recordings and fixation
- Prepare stock solutions of 10x concentrated cutting solution (125 mM NaCl, 25 mM sucrose, 2.5 mM KCl, 25 mM NaHCO3, 1.25 mM NaH2PO4, and 2.5 mM D-glucose) and artificial cerebrospinal fluid (ACSF) solution (124 mM NaCl, 2.5 mM KCl, 26 mM NaHCO3, 1 mM NaH2PO4, and 11 mM D-glucose) in pure deionized water prior to electrophysiology experiments. Store these solutions at 4 °C in 1 L bottles without CaCl2 and MgCl2.
- On the day of the experiment, dilute the stock solutions of cutting solution and ACSF 10x to a final volume of 0.5 L each. Agitate with a magnetic stirrer and oxygenize by bubbling with 95%/5% O2/CO2. Add divalent ions to obtain final concentrations of 0.1 mM CaCl2 and 7 mM MgCl2 for the cutting solution, and 2 mM CaCl2 and 2 mM MgCl2 for ACSF.
- Prepare the potassium-gluconate based pipette solution to contain: 135 mM K-gluconate, 1.2 mM KCl, 10 mM HEPES, 0.2 mM EGTA, 2 mM MgCl2, 4 mM MgATP, 0.4 mM Tris-GTP, 10 mM Na2-phosphocreatine, and 2.7–7.1 mM biocytin for post-hoc cell morphology revelation. Adjust the solution’s pH to 7.3 and osmolarity to 290 mOsm. Store 1 mL aliquots at -20 °C.
- Prepare 0.1 M PBS by diluting BupH PBS dry-blend powder pouches in 500 mL of distilled water, resulting in 0.1 M sodium phosphate, 0.15 M NaCl, pH 7.2.
- To prepare 1 L of 4% PFA solution, dilute 111 mL of 36% liquid PFA and 90 mL of 10x PBS solution in distilled water.
- Prepare 30% sucrose solution containing 150 g of sucrose in 500 mL of 0.1 M PBS.
4. Preparation of brain slices
- Prepare the bench space with absorbent bench paper before perfusion.
- Install a drip about 1 m above the bench for gravity-fed perfusion. Attach a 24 G butterfly needle.
- Surround the cutting chamber of the vibratome with ice and store it in a freezer.
- Anesthetize the mouse with intraperitoneal injection of the same ketamine-xylazine mixture used for surgery. Assess the stage of the anesthesia by pinching the toe with forceps. When fully asleep, inject 100 µL of heparin (5000 U.I./mL) intraperitoneally.
- Fix the animal with adhesive tape on the absorbent paper, lying on its back. Open the thoracic cage by cutting the ribs on the left and right sides with small scissors, from the diaphragm upwards. Maintain the thoracic cage open with the help of adhesive tape.
- Clamp the descending aorta with a hemostat and perfuse via the left ventricle of the heart with 4 °C cooled and oxygenated (95%/5% O2/CO2), cutting the solution through the 24 G butterfly needle. After 5 s, open the right atrium with small scissors.
- After 5 min of perfusion, when the organs are bloodless, stop the perfusion. Decapitate the animal with big scissors and immerge the head into 4 °C cooled and oxygenated cutting solution in a Petri dish.
- To extract the brain, cut the skin from neck to nose, then section the last vertebrae from the skull with scissors. Manually retract the skin and use small scissors to open the skull, cutting it along the midline, from caudal to rostral, upward to between the eyes.
- Carefully remove the parietal bone and caudal part of frontal bone with curved or bone forceps. Extract the brain with a small rounded spatula by inserting the instrument between the brain and the cranial floor, sectioning the olfactory bulb, optic nerve and other cranial nerves, and cerebellum.
- Gently submerge the brain in ice-cold cutting solution (4 °C) in a beaker. Position the brain on filter paper to gently dry the cortical surface. Glue the brain cortex-down to the specimen holder of a vibratome, with the caudal side facing the blade, in order to cut horizontal brain slices.
- Fill the cutting chamber with ice-cold oxygenated cutting solution so the brain is fully immerged. Make a cut on the left hemisphere (contralateral to the injected side) to avoid potential left-right ambiguity on slices.
CAUTION: Always oxygenate the solution and protect slices from light exposure. - Cut 300 µm thick slices with the vibratome, at a speed of 0.07 mm/s at 1 mm amplitude. At this stage, it is recommended to briefly check the Chronos-GFP expression in the thalamus using a fluorescent flashlight (440-460 nm) and corresponding filter glasses (500 nm long pass).
- Isolate the hippocampal region with a scalpel and transfer it to a chamber positioned in a beaker filled with bath-warmed (34 °C), oxygenated (95%/5% O2/CO2) ACSF.
- After 15 min, take the chamber out of the heated water bath and let the slices rest at room temperature, still oxygenated for at least 45 min until use.
5. Whole-cell patch-clamp recording
- Gently transfer a brain slice containing the hippocampal complex with a custom-made glass transfer pipette to the recording chamber mounted on an upright microscope. A transfer pipette is made of a shortened Pasteur pipette (inner diameter 6.5 mm) attached to a rubber pipette bulb. Continuously perfuse the recording chamber (3 mL) with 34 °C (warmed) ACSF bubbled with 95%/5% O2/CO2. Set the speed of the peristaltic pump to 2-3 mL/min.
- Briefly examine Chronos-GFP expression in axon terminals in the region of interest with blue LED illumination (470 nm) and observe with a 4x objective. GFP fluorescence is visualized through an appropriate emission filter, with a CCD camera image displayed on a computer screen.
- Place a slice anchor made from a U-shaped platinum wire with tightly spaced nylon strings ("harp") on the slice to maintain it.
- Change to a 63x immersion objective and adjust the focus. Check for axons expressing Chronos-GFP and choose a pyramidal neuron for patch recording.
- Move the objective upward.
- Pull pipettes using a Brown-Flaming electrode puller from borosilicate glass. The puller is set to produce pipettes with approximately 1 μm in tip diameter. Fill the pipettes with K-gluconate-based internal solution.
- Mount the pipette in the pipette holder on the head-stage. Lower the pipette in the chamber and find the tip under the objective. Pipette resistance should be between 3–8 MΩ. Apply a light positive pressure with a syringe so as to see a cone of solution outflow out of the pipette and progressively lower the pipette and objective to the surface of the slice.
- Patch the cell in voltage-clamp configuration: approach the identified neuron and delicately press the pipette tip onto the soma. The positive pressure should produce a dimple on the membrane surface. Release the pressure to create a giga-ohm seal (>1 GΩ resistance). Once sealed, set the holding voltage to -65 mV. Break the membrane with a sharp pulse of negative pressure: this is achieved by applying strong suction to a tube connected to the pipette holder.
- Record in whole-cell current clamp mode the responses of the neuron to hyperpolarizing and depolarizing current steps (Figure 2A).
NOTE: This protocol will be used to determine active and passive intrinsic properties of the cell. Custom-written MATLAB routines are used for off-line analysis10,16. - Record in current- or voltage-clamp postsynaptic responses to whole-field 475 nm LED stimulation of afferent fibers expressing Chronos. Stimulate with trains of 10 stimulations of 2 ms durations at 20 Hz (Figure 2B,C). Light intensity may vary from 0.1–2 mW.
NOTE: Light intensity was measured with a digital handheld optical power console equipped with a photodiode sensor, positioned under the objective. Response latencies of 2–4 ms are characteristic for a monosynaptic connection. - To investigate the nature of the synaptic transmission between the long-range afferents and the recorded neuron, different pharmacological agents may be used. To pharmacologically distinguish direct, monosynaptic responses from indirect responses via network activation, add 1 µM TTX and 100 µM 4-AP to the ACSF.
NOTE: Bath application of glutamate receptor blockers allows to determine the nature of the neurotransmitter that is released and the identity of postsynaptic receptors. For example, AMPA type glutamate receptors will be blocked by NBQX (10 µM) and NMDA receptors by APV (100 µM). Depending on the aim of the study, protocols may be conceived to investigate voltage dependence of synaptic responses or response dynamics over time. - Wash with original ACSF solution to patch another cell, or transfer the slice containing a biocytin-filled neuron in a small vial filled with 4% PFA.
- After overnight fixation in 4% PFA, wash the slice in 0.1 M PBS (2x for 5 min, 1x for 20 min).
- Store in 30% sucrose at 4 °C.
6. Biocytin revelation
- Transfer the fixed slices containing biocytin-filled neurons onto a glass blade in a drop of 30% sucrose and perform three cycles of freezing-thawing: place the blade onto dry ice disposed in a styrofoam box for 1 min until drops of sucrose are completely frozen, then press the blade against the hand palm to thaw.
- Wash the slice 3x in 0.1 M PBS (2x for 5 min, 1x for 1 h and 50 min), gently agitated. Do not exceed 2 h for the last washing.
- Pre-incubate the slice at RT for 2 h in agitated buffer solution containing 2% milk powder (0.4 g in 20 mL) to saturate non-specific sites and 0.5% Triton X100 (0.1 mL in 20 mL) to permeabilize the membranes in 0.1 M PBS.
- Incubate overnight at 4 °C in a solution containing 2% milk powder, 1% Triton X100, streptavidin-Cy5 conjugate (1/500), and DAPI (1/1000) in 0.1 M PBS, gently agitated.
- Wash the slice three times in 0.1 M PBS (2x for 5 min, 1x for 2 h). The last wash can last longer, up to 4 h, to reduce background staining.
- Before mounting the slice, use an epifluorescence microscope at 10x magnification configured to observe Cy5 fluorescent markers in order to identify the side of the slice containing the marked cell in a chamber filled with PBS.
- Transfer the slice onto a blade, cell-side up, dry it with a paper tissue, and mount it using high-resistance mounting medium.
- Use an epifluorescence microscope at 10x magnification in Cy5 and DAPI configuration to examine the cell body location, and in GFP configuration to observe the marked afferents, or a high-resolution confocal microscope at 20x for detailed somatic, axonal, and dendritic morphology (Figure 2D,E). Filter settings are detailed in the Table of Materials.
Representative Results
The procedure presented here was used to express a blue light-sensitive channelrhodopsin (Chronos) fused to GFP in the antero-dorsal nucleus of the thalamus (ADN), by stereotaxic injection of anterograde adeno-associated virus. The stereotaxic coordinates were determined according to a mouse brain atlas and tested by injecting 200 nL of fluorescent tracer fluoro-ruby. The animal was sacrificed 10 min after the injection, and the brain was extracted and fixated overnight. Coronal brain sections were prepared to examine the injection site, which was correctly placed in and limited to ADN (Figure 1A,B).
In order to express Chronos-GFP in neurons of ADN, we injected 300 nL of AAV5.Syn.Chronos-GFP.WPRE.bGH. Three weeks after the injection, acute horizontal brain slices were prepared. Figure 1C shows a brain slice containing the thalamic injection site in the right hemisphere, with GFP expression in green. Upon inspection with an epi-fluorescence microscope equipped with a 4x objective, GFP labeled thalamic axons were observed in the presubiculum (Figure 1C,D). It was noted that thalamic axons densely innervated the superficial layers I and III of the presubiculum (Figure 1D).
The activity of presubicular neurons in layer III was recorded in the whole-cell patch-clamp configuration. Hyperpolarizing and depolarizing current steps were applied while recording the membrane potential variations (Figure 2A). Data was stored on a computer for later offline analysis of active and passive membrane properties. Presubicular layer III principal cells typically possessed a negative resting potential close to -63 mV and required depolarizing current injections to drive the membrane potential to firing threshold. A full description of their intrinsic properties has been published11.
Stimulating ADN axon terminals expressing Chronos-GFP elicited excitatory post-synaptic potentials (EPSPs) in presubicular layer III principal cells in current clamp mode (Figure 2B). Depending on light intensity, the EPSPs could reach action potential threshold. Postsynaptic responses were also observed in voltage-clamp mode as excitatory post-synaptic currents (EPSCs) were elicited (Figure 2C). Onset latencies of EPSCs evoked by light stimulations were short (median, 1.4 ms10), indicating a direct synaptic contact between thalamic axons and layer III presubicular neurons. Persisting EPSCs in TTX-4AP condition confirmed this monosynaptic activation. It is noteworthy that these cells responded reliably to the light stimulations of afferent axons with a regular firing pattern.
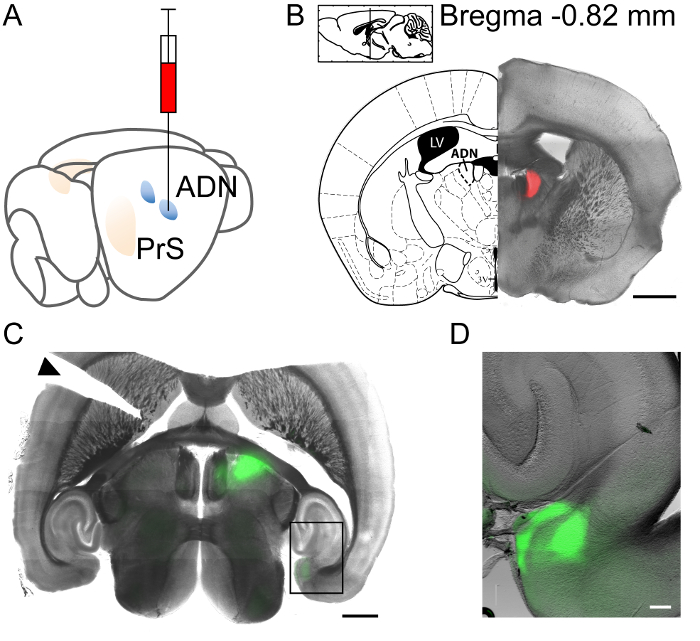
Figure 1: Stereotaxic injection in the anterodorsal thalamic nucleus (ADN). (A) Schematic representation of the injection. (B) Injection site confirmation with fluoro-ruby in a coronal section. Inset indicates antero-dorsal level and distance from bregma. (C) Horizontal slice following AAV-Chronos-GFP injection in the thalamus. The axonal projections to the ipsilateral presubiculum should be noted. An incision on the left side of the slice (indicated by a black triangle) marks the contralateral hemisphere. (B, C) Scale bar 1 mm. (D) Magnified view of inset in (C) with ADN projections to the presubicular superficial layers. Scale bar = 100 µm. Please click here to view a larger version of this figure.
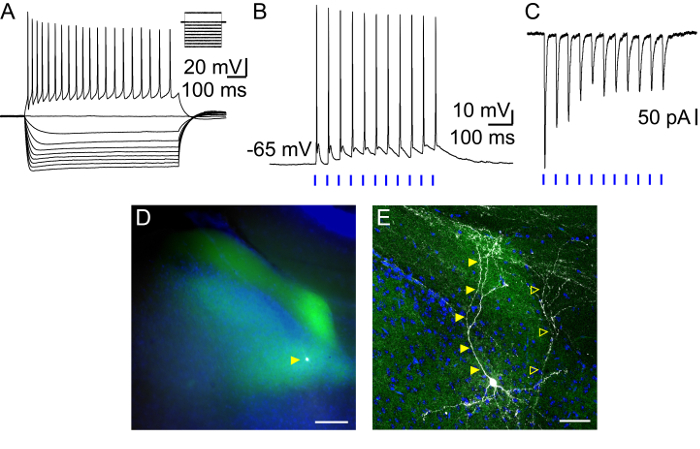
Figure 2: Presubicular layer III neuron: intrinsic properties, response to light stimulation of thalamic afferents, and post-hoc revelation of cell morphology. (A) Firing pattern and membrane potential variations of layer III neuron for hyperpolarizing and depolarizing current steps. (B, C) Responses of layer III neuron to 2 ms light stimulations (blue bars) of thalamic axons recorded in (B) current-clamp and (C) voltage-clamp modes. (D, E) Layer III pyramidal neuron (white, indicated by filled yellow triangle) surrounded by thalamic axons expressing Chronos-GFP (green) in presubicular superficial layers with DAPI staining (blue) in horizontal slice imaged with an epifluorescence microscope (D, scale bar = 100 µm) and confocal microscope at a high magnification (E, scale bar = 50 µm). The cell in (A) is indicated with filled yellow triangles. A second, partially filled neuron is present in this slice indicated with empty yellow triangles. Please click here to view a larger version of this figure.
Discussion
In vivo viral injection to express light-sensitive opsins in a defined brain area is a choice method for the optogenetic analysis of long-range functional connectivity10,11,17,18. Stereotaxic injections offer the possibility to precisely target a specific area of the brain. The coexpression of an opsin with a fluorescent reporter conveniently allows evaluation of the successful expression and confirmation of the precise injection site. The use of AAV serotype 2/5 typically restricts expression to the targeted brain region. In this way, a restricted population of neurons is transfected, expressing light-sensitive ion channels in their cell bodies and axon terminals. In subsequent ex vivo slice experiments, it is possible to stimulate these axon terminals with light pulses directly in their target area, while reading out successful synaptic transmission via patch-clamp recording of a post-synaptically connected neuron. The above protocol is robust and convenient, and some additional notes may help performance of successful experiments.
Different types of anesthesia may be used. Described here is the intraperitoneal injection of a ketamine-xylazine combination as an easy-to-use, short-term anesthesia with convenient analgesia19. The depth and duration of anesthesia may vary to some extent. In some cases, it may be necessary to inject another half-dose of ketamine-xylazine during the protocol. Isoflurane anesthesia can be a good alternative to induce more quickly and better control the depth of anesthesia. Coordinates of injection sites may be determined with the help of a mouse brain atlas. In practice, coordinates need to be tested and adjusted, if necessary.
Clean working conditions are also key. It is recommended to use disposable protective gear, including gloves, a mob cap, and a lab coat. When positioning the animal in the stereotaxic frame, special attention should be paid to the comfort of the animal, which will greatly improve efficiency of the anesthesia. The body of the animal should be aligned with the head and neck. The most critical step in positioning the animal and before craniotomy is adjustment of the bregma-lambda axis. Especially when targeting deep brain structures, even a small deviation will generate errors when lowering the injection needle into the brain. In some cases, one may deliberately choose and calculate an oblique needle trajectory.
The injection volume is a determinant factor for obtaining precisely localized opsin expression. A small volume is ideal to privilege a tightly restricted transfection zone. Higher volumes may be useful to cover the full extent of a large target area. If a large area needs to be covered, such as the septum18, it may be helpful to place several small injections with a range of neighboring coordinates. The interval until the ex vivo electrophysiological recording is also critical. A minimum time for full expression is necessary. While 3 weeks seem to be an optimal delay for these experiments11, the necessary delay may vary depending on the virus, its serotype, and the distance to the postsynaptic brain region.
The approach described here is even more powerful when combined with injections in transgenic animals. Previous work has exploited different mouse lines for subtypes of GABAergic neurons, in order to specifically target either PV- or SST-expressing interneurons for patch-clamp recordings20. Simultaneous double recording of neighboring PV and pyramidal neurons or SST and pyramidal neurons then allows comparison of strengths of long-range inputs between two neuron types11. This yields results that are standardized with respect to one neuron type. This standardization is particularly important in cases where the expression levels of opsins vary between different animals or different slices.
Slice health is essential for high-quality patch-clamp recordings. Constant oxygenation of the slices is crucial, and a slow cutting speed significantly improves slice surface quality. A slice thickness of 300 µm preserves, to some extent, the microcircuit integrity in horizontal presubicular sections, including pyramidal neurons with their cell bodies, dendritic and local axonal ramifications, and local synaptic connections. The type of light-gated channels chosen to induce activation of afferent fibers will greatly influence the stimulation parameters (duration, light intensity). Chronos is a blue light-sensitive channelrhodopsin, and a broad range of illumination wavelengths can be used for activation (peak sensitivity around 500 nm, even with minimal light intensity of 0.05 mW/mm2, also activated at 405 nm, and up to 530 nm21). Furthermore, Chronos has fast kinetics properties in comparison to classical ChR2, which enables high frequency stimulations and reliable activation of long-range projections22. In combination with the expression of Chrimson, a red-shifted opsin variant, the independent optical excitation of distinct neural populations becomes feasible.
Divulgations
The authors have nothing to disclose.
Acknowledgements
We thank Bertrand Mathon, Mérie Nassar, Li-Wen Huang, and Jean Simonnet for their help in the development of previous versions of the stereotaxic injection protocol and Marin Manuel and Patrice Jegouzo for technical help. This work was supported by the French Ministry for Education and Research (L. R., L. S.), Centre National des Etudes Spatiales (M. B.), and Agence Nationale de la Recherche Grant ANR-18-CE92-0051-01 (D. F.).
Materials
| 0.5 mm bur | Harvard Apparatus | 724962 | |
| 10 µL Hamilton syringe | Hamilton | 1701 RN – 7653-01 | |
| 10X PBS solution | Thermofisher Scientific | AM9624 | text |
| 36% PFA | Sigma-Aldrich | F8775 | |
| 470 nm LED | Cairn Research | P1105/470/LED DC/59022m | use with matched excitation filter 470/40x and emission filter for GFP |
| AAV5.Syn.Chronos-GFP.WPRE.bGH | Penn Vector Core | AV-5-PV3446 | lot V6026R, qTiter GC/ml 4.912e12, ddTiter GC/ml 2.456e13 |
| All chemicals | Sigma | ||
| Bath temperature controler | Luigs & Neumann | SM7 | Set at 34°C |
| beveled metal needle | Hamilton | 7803-05 | 33 gauge, 13mm, point style 4-20° |
| Big scissors | Dahle Allround | 50038 | |
| Biocytin | Sigma | B4261 | final 1-3 mg/ml |
| Borosilicate Capillaries | Havard Apparatus | GC150-10 | 1.5 mm outer, 0.86 inner diameter |
| Brown Flaming electrode puller | Sutter Instruments | P-87 | |
| BupH Phosphate Buffered Saline pack | Thermofisher Scientific | 28372 | |
| butterfly needle for perfusion | Braun | Venofix A | 24G |
| CCD Camera | Andor | DL-604M | |
| Confocal Microscope | Zeiss | LSM710 | 20X |
| curved forceps | FST | 11011-17 | |
| CY5 configuration (confocal) | Helium-Neon 633nm (5,0 mW) laser; Mirror: MBS 488/561/633 | ||
| CY5 configuration (epifluo) | Nikon/Chroma | Fluorescent light (Intensilight); Excitation filter: BP645/30; Dichroic mirror: 89100 BS ; Emission filter: BP705/72 | |
| DAPI | Sigma | D9542 | |
| DAPI configuration (epifluo) | Nikon/Chroma | Fluorescent light (Intensilight); Cube: Semrock Set DAPI-5060C-000-ZERO (Excitation: BP 377/50; Mirror: BS 409; Emission: BP 447/60) | |
| Digidata 1440A | Axon Instruments | ||
| Digital handheld optical meter | ThorLabs | PM100D | Parametered on 475 nm |
| Double egde stainless steel razor blades | Electron Microscopy Sciences | 72000 | Use half of the blade in the slicer |
| Dual Fluorescent Protein Flashlight | Nightsea | DFP-1 | excitation, 440-460 nm; emission filter on glasses, 500 nm longpass. |
| EGTA | Sigma | E4368 | final 0,2 mM |
| Epifluorescence Microscope | Nikon | Eclipse TE-2000E | 10 or 20X |
| Filter paper | Whatman | ||
| Fluoro-Ruby 10% | Millipore | AG335 | disolve 10 mg in 100 µl of distilled water ; inject 150 to 300 nl |
| GFP configuration (epifluo) | Nikon/Chroma | Fluorescent light (Intensilight); Cube: Filter Set Nikon B-2E/C FITC (Excitation: BP 465-495; Mirror: BS 505; Emission: BP 515-555) | |
| Heatingplate | Physitemp | HP4M | |
| Heparin choay 5000 U.I./ml | Sanofi | 5 ml vial | |
| HEPES | Sigma | H3375 | final 10 mM |
| High speed rotary micromotor kit | Foredom | K.1070 | maximum drill speed 38,000 rpm |
| Internal solution compounds : | |||
| Isolated Pulse Stimulator | A-M Systems | 2100 | |
| KCl | Sigma | P4504 | final 1,2 mM |
| Ketamine 1000 | Virbac | ||
| Ketofen 10% | Merial | 100 mg/ml : dilute 1 µl in 1ml total (0,1%) | |
| Laocaine (lidocaine) | MSD | 16,22 mg/ml : dilute 1 ml in 4 ml total (around 4%) | |
| LED hi power spot for surgery | Photonic (via Phymep) | 10044 | |
| LED Power Supply | Cairn Research | OptoLED Light Source | |
| Manipulators | Luigs & Neumann | SM-7 | |
| Mg-ATP 2H20 | Sigma | A9187 | final 4 mM |
| MgCl2 | Sigma | 63069 | final 2 mM |
| Micro temperature controler | Physitemp | MTC-1 | |
| Milk powder | Carnation | ||
| MultiClamp 700B | Axon Instruments | ||
| Na Phosphocreatine | Sigma | P7936 | final 10 mM |
| Na3-GTP 2H20 | Sigma | G9002 | final 0.4 mM |
| needle holder/hemostat | FST | 13005-14 | |
| pClamp acquisition software | Axon Instruments | ||
| Peristaltic pump | Gilson | Minipuls 3 | 14-16 on the display for 2-3 ml/min |
| Potassium gluconate (K-gluconate) | Sigma | G4500 | Final 135 mM |
| ProLong Gold antifade mounting medium | Thermofisher Scientific | P36390 | |
| Rompun 2% (xylazine) | Bayer | ||
| small scissors | FST | 14060-09 | |
| Sodium chloride 0.9% | Virbac | dilute 8.5 mL in 10 ml total | |
| Stereomicroscope VISISCOPE SZT | VWR | 630-1584 | |
| Stereotaxic frame with digital display | Kopf | Model 940 | Small animal stereotaxic instrument |
| Streptavidin-Cy3 conjugate | Life technologies | 434315 | |
| Streptavidin-Cy5 conjugate | Thermofisher Scientific | S32357 | |
| Superglue3 Loctite | Dutscher | 999227 | 1g tube |
| Suture filament Ethilon II 4-0 polyamid | Ethicon | F3210 | |
| Syringe pump | kdScientific | Legato 130 – 788130 | Use Infuse and Withdraw modes |
| Tissue slicer | Leica | VT1200S | speed 0.07, amplitude 1. |
| tubing | Gilson | F117942, F117946 | Yellow/Black, Purple/Black |
| upright microscope | Olympus | BX51W1 | |
| Versi-dry bench absorbant paper | Nalgene |
References
- Goodridge, J. P., Taube, J. S. Interaction between the postsubiculum and anterior thalamus in the generation of head direction cell activity. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 17 (23), 9315-9330 (1997).
- Winter, S. S., Clark, B. J., Taube, J. S. Spatial navigation. Disruption of the head direction cell network impairs the parahippocampal grid cell signal. Science. 347 (6224), 870-874 (2015).
- Fan, Y., et al. Activity-dependent decrease of excitability in rat hippocampal neurons through increases in I(h). Nature Neuroscience. 8 (11), 1542-1551 (2005).
- Takahashi, H., Magee, J. C. Pathway Interactions and Synaptic Plasticity in the Dendritic Tuft Regions of CA1 Pyramidal Neurons. Neuron. 62 (1), 102-111 (2009).
- Dolleman-van der Weel, M. J., Lopes da Silva, F. H., Witter, M. P. Interaction of nucleus reuniens and entorhinal cortex projections in hippocampal field CA1 of the rat. Brain Structure & Function. 222 (5), 2421-2438 (2017).
- Callaway, E. M., Yuste, R. Stimulating neurons with light. Current Opinion in Neurobiology. 12 (5), 587-592 (2002).
- Fino, E., et al. RuBi-Glutamate: Two-Photon and Visible-Light Photoactivation of Neurons and Dendritic spines. Frontiers in Neural Circuits. 3, 2 (2009).
- Mao, T., et al. Long-range neuronal circuits underlying the interaction between sensory and motor cortex. Neuron. 72 (1), 111-123 (2011).
- Zhang, F., et al. Optogenetic interrogation of neural circuits: technology for probing mammalian brain structures. Nature Protocols. 5 (3), 439-456 (2010).
- Simonnet, J., et al. Activity dependent feedback inhibition may maintain head direction signals in mouse presubiculum. Nature Communications. 8, 16032 (2017).
- Nassar, M., et al. Anterior Thalamic Excitation and Feedforward Inhibition of Presubicular Neurons Projecting to Medial Entorhinal Cortex. Journal of Neuroscience. 38 (28), 6411-6425 (2018).
- Fricker, D., et al. Pyramidal cells of rodent presubiculum express a tetrodotoxin-insensitive Na+ current. The Journal of Physiology. 587, 4249-4264 (2009).
- Simonnet, J., Eugène, E., Cohen, I., Miles, R., Fricker, D. Cellular neuroanatomy of rat presubiculum. The European Journal of Neuroscience. 37 (4), 583-597 (2013).
- Simonnet, J., Fricker, D. Cellular components and circuitry of the presubiculum and its functional role in the head direction system. Cell and Tissue Research. 373 (3), 541-556 (2018).
- Paxinos, G., Franklin, K. B. J. . The Mouse Brain in Stereotaxic Coordinates. , (2013).
- Huang, L. -. W., et al. Laminar Localization and Projection-Specific Properties of Presubicular Neurons Targeting the Lateral Mammillary Nucleus, Thalamus, or Medial Entorhinal Cortex. eNeuro. 4 (2), (2017).
- Cruikshank, S. J., Urabe, H., Nurmikko, A. V., Connors, B. W. Pathway-specific feedforward circuits between thalamus and neocortex revealed by selective optical stimulation of axons. Neuron. 65 (2), 230-245 (2010).
- Gonzalez-Sulser, A., et al. GABAergic Projections from the Medial Septum Selectively Inhibit Interneurons in the Medial Entorhinal Cortex. Journal of Neuroscience. 34 (50), 16739-16743 (2014).
- Mathon, B., et al. Increasing the effectiveness of intracerebral injections in adult and neonatal mice: a neurosurgical point of view. Neuroscience Bulletin. 31 (6), 685-696 (2015).
- Nassar, M., et al. Diversity and overlap of parvalbumin and somatostatin expressing interneurons in mouse presubiculum. Frontiers in Neural Circuits. 9, 20 (2015).
- Klapoetke, N. C., et al. Independent optical excitation of distinct neural populations. Nature Methods. 11 (3), 338-346 (2014).
- Hass, C. A., Glickfeld, L. L. High-fidelity optical excitation of cortico-cortical projections at physiological frequencies. Journal of Neurophysiology. 116 (5), 2056-2066 (2016).
