All experimental animal procedures were conducted with 200 µm, fixed, sectioned mouse brains, in accordance with the protocol (# 4395-01) approved by the Institute of Animal Care and Use Committee (IACUC) of the University of Washington. Wild-type mice (C57BL/6J strain) are euthanized with CO2. Then, a craniotomy is performed to extract their brains for fixation in 4% paraformaldehyde in phosphate-buffered saline. The brains are embedded in a 3% agarose and 0.3% gelatin mixture and sectioned into 200 μm-thick slices by a vibratome.
1. Initial alignment
NOTE: Ensure that the beam size and divergence of both arms are matched for best sensitivity and resolution. Collimate the pump and the Stokes beams and adjust their sizes before they enter the laser scanning microscope. To do this, use a pair of achromatic lenses for each beam before combining them on the dichroic mirror. Always wear proper laser goggles for beam alignment.
- Beam collimation
- Install a pair of achromatic lenses for the pump beam. As a starting point, use a 100 mm lens and 200 mm lens to magnify the laser beam size by 2-fold. Ensure that the distance of the two lenses is roughly 300 mm. Align the pump beam through the center of both lenses.
- Place a mirror after the second lens to send the beam toward a wall (>1 m away). Take care when sending the beam across long distances. Trace the beam from the mirror to the wall with an IR card and check if the beam changes in size. Collimate the beam if the beam changes in size as a function of distance.
- If the beam is converging (decreasing size with propagation), move the two lenses closer.
- If the beam is diverging (increasing size with propagation), move the two lenses further apart. Adjust the distance until the beam is collimated.
- Repeat steps 1.1.1 and 1.1.2 for the Stokes beam to collimate the beam.
- Beam size adjustment
- If a beam profiler is available, measure the collimated beam size for each beam. Alternatively, estimate the beam size using the IR card and a ruler to obtain a beam diameter of 4-5 mm.
- If the beam size is too small or too large, change the lens pair used in step 1.1. Adjust the lens pair until both beams have a diameter of 4-5 mm.
NOTE: The magnification of the beam is the ratio between the focal length of the second lens to the first lens (f2/f1).
- Spatial overlap
NOTE: SRS imaging requires both laser beams to be combined in space and time to excite molecular vibrations. A schematic of the spectral-focusing SRS imaging is shown in Figure 1.- Combine the two laser beams by installing a dichroic mirror with several steering mirrors for adjustment. Optimize spatial overlap of the pump and Stokes by monitoring beams after the dichroic mirror at two different positions far apart (~1 m). Iteratively adjust the steering mirror before the dichroic and the dichroic mirror to align the Stokes beam with the pump beam.
NOTE: If the two beams are spatially overlapping at both positions, they are sufficiently overlapped. - Ensure that the combined beams are sent to the center of the scan mirrors of the laser scanning microscope by adjusting a pair of steering mirrors when the scan mirrors are in the parked position. Ensure both beams travel through the center of the microscope objective and the condenser.
- After the condenser, use another pair of lenses with focal lengths of 100 mm and 30 mm, respectively, to relay the transmitted beam onto the photodiode. Ensure both beams are contained within the photodiode and install two low-pass filters to block out the modulated Stokes beam.
- Combine the two laser beams by installing a dichroic mirror with several steering mirrors for adjustment. Optimize spatial overlap of the pump and Stokes by monitoring beams after the dichroic mirror at two different positions far apart (~1 m). Iteratively adjust the steering mirror before the dichroic and the dichroic mirror to align the Stokes beam with the pump beam.
2. SRS signal detection
- Electrooptical modulation (EOM)
NOTE: EOM of 20 MHz is used to modulate the Stokes amplitude. As discussed later, the EOM is derived from the 80 MHz laser pulse train, which is required for orthogonal modulation. Other modulation frequencies can be used if only single-color or hyperspectral SRS is performed. In that case, synchronization of modulation frequency to laser frequency is unnecessary. A frequency generator with an RF power amplifier can be used to drive the EOM. As a result, steps 2.1.1-2.1.4 can be skipped.- Place a beam sampler in the Stokes beam path to pick up 10% of the beam and send it to a fast photodiode to detect the 80 MHz pulse train.
NOTE: The photodiode signal is sent to a frequency divider to generate a 20 MHz TTL output. This output is further sent into a fanout buffer to replicate the output into four identical 20 MHz outputs. One of the outputs is used to trigger the oscilloscope. - Take one of the outputs of the fanout buffer and filter it with a bandpass filter to get a 20 MHz sinusoidal wave. Use an RF attenuator to adjust the output peak-to-peak voltage to ~500 mV.
- Send the resulting output to a phase shifter, which allows fine adjustment of the RF phase with a voltage source. Send this output to an RF power amplifier and connect the output of the amplifier to the EOM.
- Unblock the Stokes beam and optimize the modulation depth of EOM1 by placing a photodiode in the beam path. Adjust the EOM voltage and quarter-wave plate until the modulation depth (valley to peak ratio) appears satisfactory.
NOTE: At 20 MHz modulation (1/4 of the laser repetition rate), two pulses are expected every 50 ns.
- Place a beam sampler in the Stokes beam path to pick up 10% of the beam and send it to a fast photodiode to detect the 80 MHz pulse train.
- Temporal overlap
NOTE: Temporal overlap of the pump and Stokes is achieved by delaying one of the two laser pulse trains with a retro-reflector mounted onto a delay stage (Figure 1 shows the Stokes being delayed). Coarse overlap is monitored with the oscilloscope, and fine overlap is monitored by the SRS signal. Fine temporal overlap can also be achieved with an autocorrelator if available.- Place a photodiode after the dichroic mirror to detect the laser beam. Block the Stokes beam first. Zoom in on one of the pump pulse peaks on the oscilloscope. Place a vertical cursor to mark the temporal position of this peak with the oscilloscope.
- Block the pump beam and unblock the Stokes beam. Translate the delay stage to temporally match the peak position on the oscilloscope to the marked position in the previous step. See Figure 2 for a display of the temporal overlap of two beams.
- (OPTIONAL) If the translation of the delay stage is insufficient to temporally match the two beams, then move the delay stage to the middle of its movement range.
- Calculate the delay distance required to match the two beams by taking the temporal difference between the two beams and multiplying the difference by the speed of light to find the amount of distance needed to match the two beams temporally.
- Elongate the beam path of the faster beam or shorten the beam path of the slower beam to roughly match the temporal delay accordingly.
- Prepare a microscope slide sample with DMSO and double-sided tape as a spacer to hold the sample between the slide and a coverslip.
- Place the sample on the microscope with the coverslip side facing the microscope objective. Change the microscope to brightfield illumination and observe the sample from the eyepiece. Find the focus of the sample by first finding the focus at both the top and bottom layer of air bubbles at the glass-tape interface, and then move the focus to be in between the two layers of tape.
NOTE: Ensure the laser beams are blocked before looking into the eyepiece. - Set the tunable beam output to 798 nm. Based on the optical throughput of the condenser, adjust the optical power to be ~40 mW each for the pump and Stokes beams at the objective focus.
- Open ScanImage in MATLAB (or other scanning software that controls the microscope) and click on the button labeled FOCUS to start scanning.
NOTE: The laser beams will be raster-scanned through the sample to generate an image. The low-frequency signal output from the photodiode (<100 kHz) is directly sent into channel 1 of the data acquisition card (referred to as the DC channel). The high-frequency output (>100 kHz) from the photodiode is sent into the lock-in amplifier, and the X-output of the lock-in amplifier signal is sent into channel 2 of the data acquisition card (referred to as the AC channel). - Adjust the steering mirror before the galvo scanner to center the DC signal on the channel 1 display. Move the motorized delay stage and closely observe the lock-in output shown on the channel 2 (i.e., AC channel) display.
NOTE: When the pump and Stokes coincide in time, a signal will show up on the AC channel. It is helpful to adjust the color scale of the AC channel to display the small intensity change. - Maximize the AC signal intensity by finely adjusting the time delay. Adjust the dichroic mirror to center the SRS signal on the AC channel (while keeping the DC channel centered). Adjust the phase of the lock-in amplifier to maximize the signal. See Figure 3 for a satisfactory signal.
3. Spectral resolution optimization
NOTE: The pump and Stokes beams reaching the sample should have the same amount of group delay dispersion (GDD) to maximize spectral resolution. The dispersion depends heavily on the experimental setup. The experimental setup described here utilizes femtosecond pulses at 1,040 nm and 800 nm as Stokes and pump, respectively. Dense flint glass rods (H-ZF52A) are used as the pulse-stretching medium.
- Insert 48 cm of a highly dispersive glass rod (H-ZF52A or equivalent dense flint glass) into the 800 nm beam path. Estimate the GDD using Eq (1):
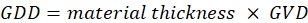 (1)
(1)
NOTE: GVD of various glass materials at different wavelengths can be found from the refractive index database resource. For example, H-ZF52A has a GVD of 220.40 fs2/mm at 800 nm. The total GDD is 105792 fs2. - Calculate how many cm of the dispersive glass rod is required to add to the 1,040 nm beam path to match the GDD of the pump. Insert the appropriate length of dispersive glass rods to the 1,040 nm beam path to roughly match the GDD of the 800 nm beam. Note that the addition of glass rods will change the temporal overlap of the two beams, and adjustment of delay may be necessary.
- Calibration of spectral resolution
- Make a microscope slide sample with DMSO. Place the slide onto the microscope and check the power of the beams coming out of the microscope condenser. Adjust the power accordingly to have ~40 mW each at sample focus.
- Open ScanImage from MATLAB. Find the maximal SRS signal by scanning through the delay stage, which corresponds to the 2,913 cm-1 Raman peak of DMSO. Estimate the stage position based on the previous stage position with the increased optical path length due to the insertion of rods. Realign the beam spatial overlap because of the small deviation of the beam when glass rods are added.
- Save a hyperspectral SRS scan by sequentially taking a series of SRS images while moving the motorized stage.
NOTE: The delay scan range covers two Raman peaks, corresponding to the 2,913 cm-1 and 2,994 cm-1 Raman peaks of DMSO, respectively. These two transitions are observed when utilizing an 800 nm pump and 1,040 nm Stokes laser. - Plot out the SRS spectra of the DMSO solution using either ImageJ or MATLAB. Fit the large DMSO 2,913 cm-1 peak to a Gaussian or Lorentzian function in MATLAB to calculate the Full Width at Half Maximum (FWHM) of the peak.
NOTE: Representative results are shown in Figure 4. If only one broad peak is present, that means either the spectral resolution is too poor to distinguish the two peaks and more glass rods are required, or the scanned range was too small to detect the second peak. Typically, an acceptable spectral resolution DMSO is ~20-25 cm-1 when glass rods length of >60 cm are used. A lower resolution is often used for tissue imaging to trade for higher signals with shorter pulses17.
- (OPTIONAL) Use an autocorrelator or a FROG (Frequency-Resolved Optical Gating) to determine the pulse duration of each arm to calculate exactly the amount of GDD and the length of rods needed to match the GDD between the pump and the Stokes.
- Repeat steps 3.3.2-3.3.4 for different rods lengths on the Stokes beam to find the optimal spectral resolution, which means the best GDD match has been found experimentally. Use multiple sets of glass rods differing in length to achieve optimal spectral resolution.
4. Signal to noise (SNR) characterization
- Ensure that step 4.2 is performed after complete spatial and temporal alignment.
- Acquire an SRS image corresponding to the 2,913 cm-1 Raman peak of DMSO. Open the image in ImageJ and select a small area in the center of the frame. Use the measure function to calculate the mean and standard deviation of values in the selected area.
- Divide the mean value of the selected area by the standard deviation to find the SNR value, as in Eq (2).
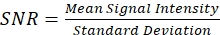 (2)
(2)
NOTE: A good SNR for the system (with a lock-in time constant of 4 μs) using DMSO at 40 mw/40 mw at focus for both arms is >800. Lower concentrations of DMSO or lower power can be used for a more accurate estimation of the SNR if the data acquisition card has a limited bit depth. - If the SNR is too low, realign the laser pulses to optimize spatial overlap, temporal overlap, beam size/collimation matching, and/or dispersion matching. For an objective with an aberration correction collar, optimize the signal by adjusting the correction collar.
5. Frequency axis calibration
NOTE: This step is performed to relate the delay stage position to the scanned Raman transition. Careful selection of solvents is required to generate an appropriate "Raman Ruler." DMSO is an effective solvent for CH bonds as it has two sharp Raman peaks at 2,913 cm-1 and 2,994 cm-1.
- Save a hyperspectral scan with the delay-stage range covering the 2,913 cm-1 and 2,994 cm-1 Raman peaks of DMSO. Save the stage positions corresponding to the hyperspectral dataset.
NOTE: The global maximum peak of the spectrum corresponds to the DMSO 2,913 cm-1 Raman shift and the second maximum peak corresponds to the DMSO 2,994 cm-1 Raman shift. - Perform linear regression for the stage positions and Raman shifts at 2,913 cm-1 and 2,994 cm-1. Using the linear regression equation relating the stage position to the Raman shift, convert the delay positions to the corresponding Raman frequencies.
6. Orthogonal modulation and two-color imaging
NOTE: The orthogonal modulation step is only necessary when real-time two-color imaging is needed. A schematic of this scheme is shown in Figure 5. The orthogonal modulation uses a pair of EOMs driven at a quarter of the laser frequency (20 MHz for 80 MHz laser) with a 90° phase shift between the two. This orthogonal modulation step can be skipped for single-color SRS imaging or hyperspectral SRS imaging.
- EOM1 modulation
- Install a PBS (PBS2), a quarter-wave plate (QWP2), and a second EOM (EOM2) into the Stokes beam path after the first EOM. Unplug the signal input to EOM2. Plug in the signal input to EOM1 and turn it on.
- Modulate the Stokes beam (fixed at 1,040 nm) at 20 MHz (f0/4) by sending the beam through the first EOM. Adjust the tilt and position of EOM1 to ensure that the beam is hitting straight and centered through the EOM crystal.
- Monitor the modulation depth by observing both polarizations coming out of PBS1 with two photodiodes and displaying the modulation on an oscilloscope.
- Adjust the QWP1, EOM1 voltage, and phase of the 20 MHz input (using a phase shifter) to optimize the modulation depth of the transmitted beam to be close to 100%. See Figure 2B for an illustration of good modulation depth.
- EOM2 modulation
- Unplug EOM1 and plug in EOM2.
- Send in the high voltage output of the second amplifier at 20 MHz to EOM2. Adjust the tilt and position of EOM2 to ensure that the beam is hitting straight and centered through the EOM crystal.
- Once again, monitor the modulation depth by looking at both polarizations coming out of PBS2 with an oscilloscope. Adjust the QWP2, EOM2 voltage, and the phase shifter as needed to achieve close to 100% modulation for both polarizations.
- Ensure that the pulse train modulation has a 90° phase shift from the first modulation.
NOTE: If the two pump pulse trains are not 90° orthogonal, crosstalk between the two channels will be a problem. - Test the orthogonality of the modulation by turning on and plugging in both EOM1 and EOM2. Monitor both polarizations being split by PBS2 with an oscilloscope. Reoptimize EOM1 and EOM2 individually if the pulse train after the second PBS does not resemble Figure 2C.
- Install a 20 mm birefringent quartz crystal (BRC) and HWP downstream of EOM2. Plug in both EOMs at once and monitor the pulse train such that it resembles Figure 2D.
NOTE: For the chirp used in this experiment, 20 mm BRC induces a time delay that corresponds to an 80 cm-1 Raman shift. A different BRC length may be needed if a different chirp is used.
- Calibration
- Calibrate the system using DMSO by detecting signals from the lock-in amplifier X and Y channel output (sent to channels 2 and 3 of the data acquisition card).
- Check whether the signals generated by the 2,913 cm-1 peak from the faster polarization and that from the slower polarization are close to 90° out-of-phase on the lock-in amplifier. If this is not the case, adjust the EOM alignment until the two signals are close to 90° out-of-phase.
- Once calibration is complete, find the delay position that probes the protein transition at 2,930 cm-1 for one of the orthogonal beams. Ensure that the other polarization probes the lipid transition of 2,850 cm-1.
7. Epi-mode SRS imaging
NOTE: In the transmission mode imaging scheme, the objective focuses the laser into the sample, and then a condenser lens directs the transmitted beam to a photodiode for lock-in detection. In the epi-mode imaging scheme, light that is backscattered and depolarized by the sample is recollected by the focusing objective and isolated using a polarizing beam splitter. The isolated and backscattered photons are sent to a photodiode through a pair of relay lenses for lock-in detection. Figure 6 depicts the epi-mode imaging scheme.
- Install an HWP before the beam enters the microscope to change the polarization of the beam going into the microscope. Place a PBS above the objective to allow the depolarized back-reflected beam to reach the detector.
- Use a pair of lenses consisting of a 75 mm achromat lens and a 30 mm aspheric lens to relay the backscattered photons from the back aperture of the objective to the photodetector. Mount the detector to collect the backscattered light directed by the PBS. Install a filter to block out the modulated beam from entering the detector.
- Place the tissue sample under the objective. As the condenser is unnecessary for epi-mode imaging, remove it if more space is required.
- Imaging
- Block off the beam with a shutter; shine a white light source onto the sample from the side; and use brightfield to find objective focus.
- Unblock both beams and use the precalibrated delay positions to acquire lipid and protein SRS images from tissue from the two outputs of the lock-in amplifier.
- Adjust the lock-in gain and pixel bin factor to acquire good-quality images.
8. False-color staining
- Open the image stack with ImageJ.
- Pull out the two images that correspond to the lipid (2,850 cm-1) and protein (2,930 cm-1) species by right-clicking on the image and clicking on Duplicate.
- Rename the lipid image to lipids and the protein image to proteins.
- Go to Process | Image Calculator and perform proteins subtract lipids.
- Combine the images by going to Image | Color | Merge Channels, setting lipids to green and proteins to blue. Open the image channels tool (Image | Color | Channels Tool) and adjust the brightness and contrast (Image | Adjust | Brightness/Contrast).
- Adjust the brightness and contrast for each channel using the channels tool. For the lipid channel, adjust the contrast until the cellular features appear dark. For the protein channel, adjust the contrast until the cellular features appear blue. Convert the merged channel green/blue image to an RGB image by going to Image | Type | RGB Color. Export this image by File | Save As | Tiff.
NOTE: For false H&E staining, the color scheme shows pink cytoplasm, while the nuclei are dark blue-purple. - Download the HE.m MATLAB script from the false H&E staining script resource in the Table of Materials.
- Run the HE.m script in MATLAB. Select the exported RGB image from the previous step to generate an artificially H&E stained image.
- (OPTIONAL) Normalize the image intensity for large field-of-view imaging because the image appears darker in the periphery than in the center.
- To perform field normalization of the images, average as many images as possible. Then, remove the intensity features with ImageJ (Process | Filters | Gaussian Blur | Radius=50).
- Measure the maximum intensity of the blurred image (Ctrl+M). Divide the blurred image by the maximum intensity (Process | Math | Divide). Divide the raw SRS image by the blurred image (Process | Image | Calculator).
Optimizing spectral resolution:
Dispersion through a material is affected by the dispersive medium (length and material) and wavelength. Changing the dispersion rod length affects the spectral resolution and the signal size. It is a give-and-take relationship that can be weighed differently depending on the application. The rods stretch out the beam pulse from being wide in frequency and narrow in time to being narrow in frequency and broad in time. Figure 7 shows the effect of varying rod lengths on the spectral resolution. Figure 7A demonstrates very poor spectral resolution; this setup has no glass chirping rods, and the two Raman peaks from DMSO are not resolved at all. In Figure 7B,C, an increase in the number of glass chirping rods starts to resolve the two peaks. Lastly, Figure 7D shows how matched chirping resolves both peaks and can be used to calibrate stage positions to frequency.
Calibration with DMSO:
DMSO has two sharp Raman peaks at 2,913 and 2,994 cm-1 that are convenient for calibration in the C-H region. Once a DMSO spectrum is obtained from a hyperspectral SRS scan, a simple linear regression converts the stage position to the Raman shift. If the spectral resolution is poor (as shown in Figure 7A) and the two peaks are not separable, then calibration with linear regression is impossible. In this case, better dispersion matching is necessary by either adding or removing glass rods. The most common difficulties in finding a DMSO signal for calibration are due to errors in either temporal overlap or spatial overlap of the two beams. Before attempting DMSO calibration, repeat the spatial and temporal alignment steps to optimize the SRS signal.
Two-color DMSO:
In two-color SRS, two pump pulse trains are generated with orthogonal polarization with a fixed time delay (due to a birefringent crystal). Evaluation of modulation depth and 90° phase shift is done with a photodiode followed by a DMSO SRS signal. Figure 8 demonstrates acceptable modulation depth and temporal separation. Although the spectral resolution of DMSO in Figure 8 is not ideal, it is often sacrificed in tissue imaging experiments to achieve a higher signal with shorter pulses. Figure 9 demonstrates poor phase shifts that give rise to inverted or negative peaks.
Two-color SRS of mouse brain tissue in epi-mode:
Epi-mode (detecting backscattered photons) SRS is used for imaging thick tissue (>1 mm). Figure 10A,B demonstrates real-time, two-color SRS imaging at 2,850 cm-1 and 2,930 cm-1 of ex vivo mouse brain tissue. The raw images from the lipid and protein channels (Figure 10A,B) were color-coded to produce a single image depicting lipid and protein contributions (Figure 10C). False H&E staining was performed (Figure 10D) on Figure 10C to mimic H&E staining. Poor imaging quality can be a result of large imaging depth or poor calibration (frequency axis or modulation depth). The typical imaging depth in brain tissue is 100-200 μm13.
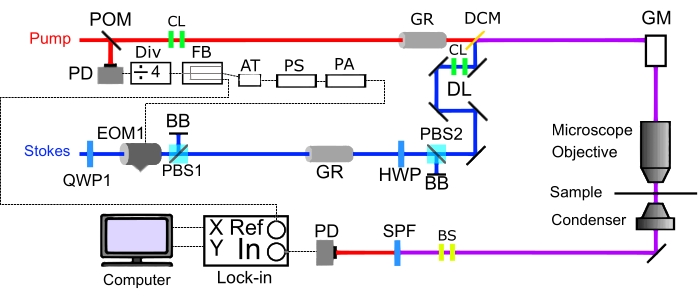
Figure 1: Schematic of one-color SRS imaging setup. Construction of a spectral-focusing SRS microscope in transmission mode. X and Y represent the orthogonal outputs. Abbreviations: SRS = stimulated Raman scattering; DL = retroreflector-based delay line; Div = divider; FB = fanout buffer; AT = attenuator; PS = phase shifter; PA = power amplifier; DCM = dichroic mirror; GM = galvo mirrors; EOM = electrooptic modulator; POM = pick-off mirror; PBS = polarizing beam splitter, BRC = birefringent crystal; QWP = quarter-wave plate; HWP: half-wave plate; PD = photodiode; GR = glass rod; BB = Beam Block; SPF = shortpass filter; CL = collimating lens; BS = beam size changing lens. Please click here to view a larger version of this figure.
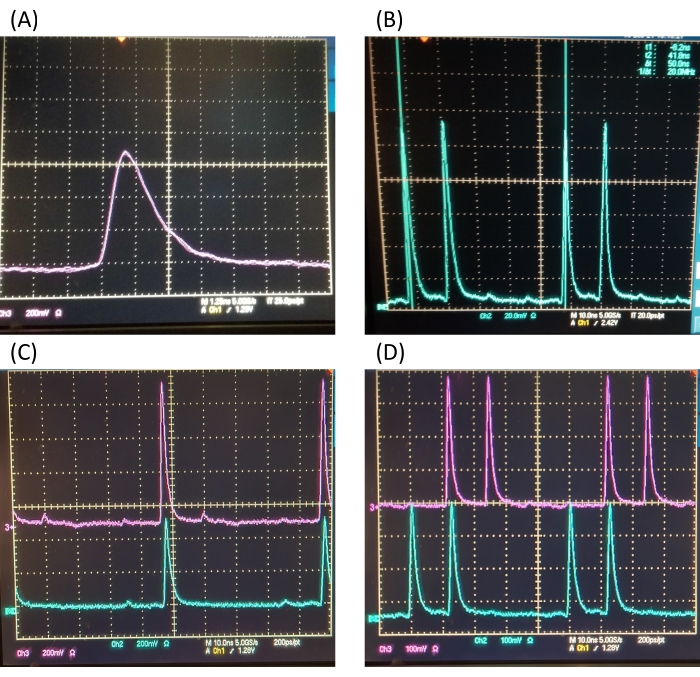
Figure 2: Representative temporal overlap. (A) The pump and Stokes beams are seen to be temporally overlapped on the oscilloscope. Oscilloscope cursors are used to mark the temporal positions of the pump and Stokes beams. This overlap is satisfactory as a starting point to further tweak temporal overlap with a delay stage. (B) Satisfactory representative modulation depth of one EOM at 20 MHz. (C) Satisfactory pulse modulation while two EOMs are in use. (D) Satisfactory pulse train modulation after birefringent quartz crystal and half-wave plate installation on the doubly modulated Stokes arm. Abbreviation: EOM = electrooptic modulator. Please click here to view a larger version of this figure.

Figure 3: Representative SRS and pump signals. (A) Misaligned pump signal detected in DC channel. (B) Misaligned SRS signal detected by photodiode. (C) Satisfactory, centered pump signal in the DC channel. (D) Satisfactory SRS signal centered on the AC channel. Abbreviation: SRS = stimulated Raman scattering. Please click here to view a larger version of this figure.
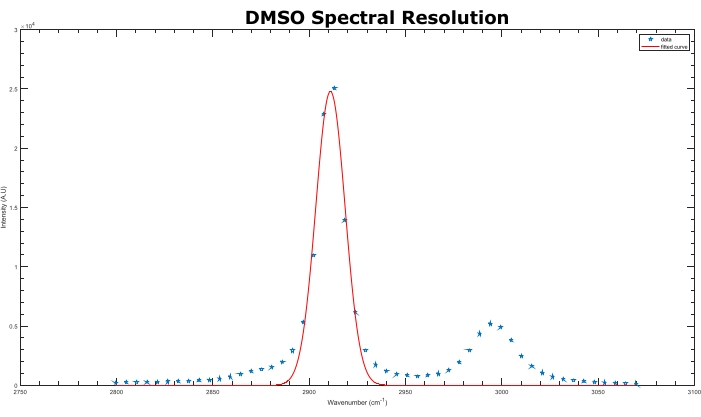
Figure 4: Spectral resolution characterization. A gaussian function was fit to the 2,913 cm-1 Raman DMSO peak. The FWHM calculated gave a resolution of 15 cm-1 for the system. Abbreviations: DMSO = dimethyl sulfoxide; FWHM = Full Width at Half Maximum. Please click here to view a larger version of this figure.
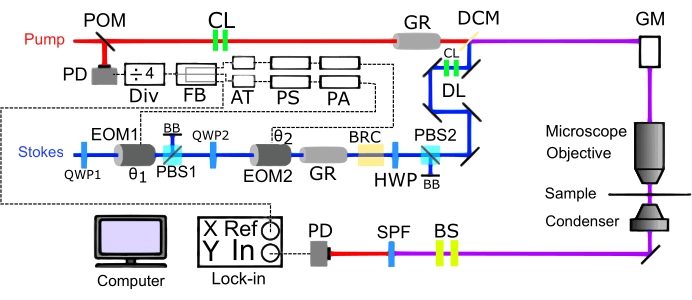
Figure 5: Schematic of two-color SRS imaging setup. Construction of a two-color SRS microscope in the transmission mode. X and Y represent the orthogonal outputs. Abbreviations: DL = retroreflector based delay line; Div = divider; FB = fanout buffer; AT = attenuator; PS = phase shifter; PA = power amplifier; DCM = dichroic mirror; GM = galvo mirrors; EOM = electrooptic modulator; PBS = polarizing beam splitter; BRC = birefringent crystal; QWP = quarter-wave plate; HWP = half-wave plate; PD = photodiode; GR = glass rod; BB = Beam Block, SPF = shortpass filter; CL = collimating lens; POM = pick-off mirror; BS = beam size changing lens. Please click here to view a larger version of this figure.
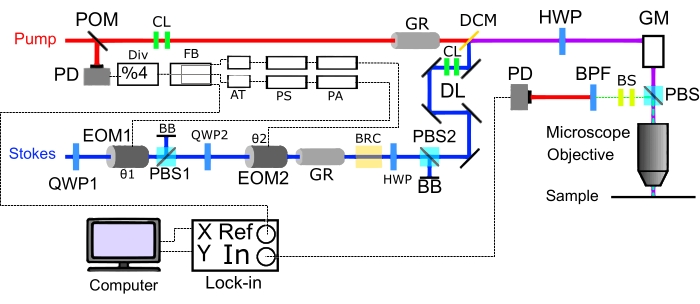
Figure 6: Schematic of two-color SRS imaging Epi-mode set-up. Construction of a two-color SRS microscope in the epi-mode. X and Y represent the orthogonal outputs. Abbreviations: DL = retroreflector based delay line; Div = divider; FB = fanout buffer; AT = attenuator; PS = phase shifter; PA = power amplifier; DCM = dichroic mirror; GM = galvo mirrors; EOM = electrooptic modulator; PBS = polarizing beam splitter; BRC = birefringent crystal; QWP = quarter-wave plate; HWP = half-wave plate; PD = photodiode; GR = glass rod; BB = Beam Block, BPF = bandpass filter; CL = collimating lens; POM = pick-off mirror; BS = beam size changing lens. Please click here to view a larger version of this figure.
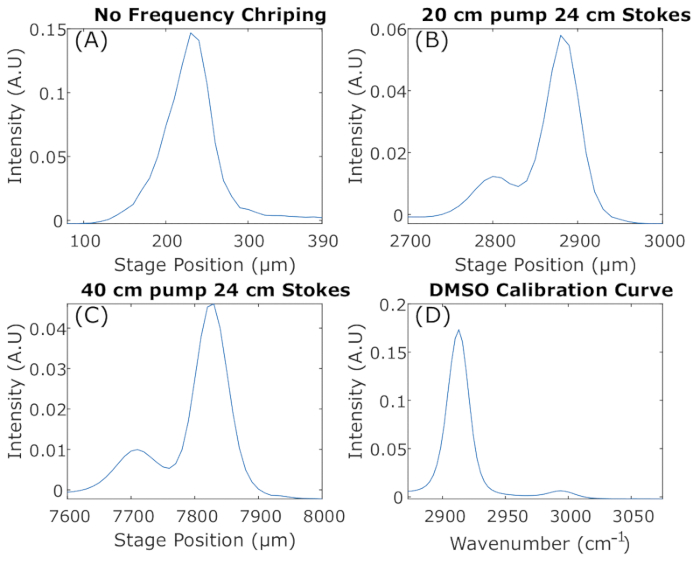
Figure 7: Optimizing spectral resolution with 25% DMSO. (A) Zero glass chirping rods were used to obtain the DMSO spectrum. The two peaks are not resolved. (B) Glass chirping rods were used, 20 cm on the pump arm and 24 cm on the Stokes arm. Two peaks are starting to be resolved to a satisfactory point. (C) Chirping rods were used, 40 cm long pump and 24 cm long Stokes, to obtain a better resolved DMSO spectrum. (D) Chirping rods were used, 64 cm long pump and 60 cm long Stokes, to obtain higher spectral resolution. Abbreviation: DMSO = dimethyl sulfoxide. Please click here to view a larger version of this figure.
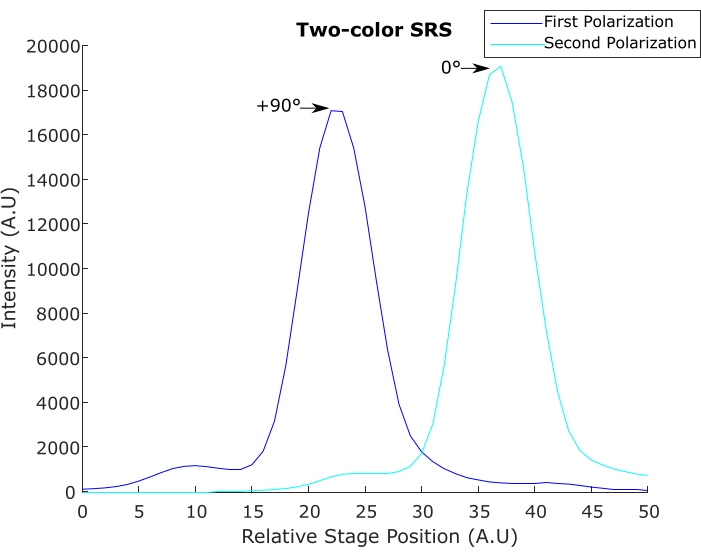
Figure 8: Two-color SRS, varying polarization and time delay. Two time-delayed pulse trains of orthogonal polarization (s&p) are used to image DMSO spectra to show the time delay (and Raman frequency difference) between the SRS excitations. Abbreviations: DMSO = dimethyl sulfoxide; SRS = stimulated Raman scattering. Please click here to view a larger version of this figure.
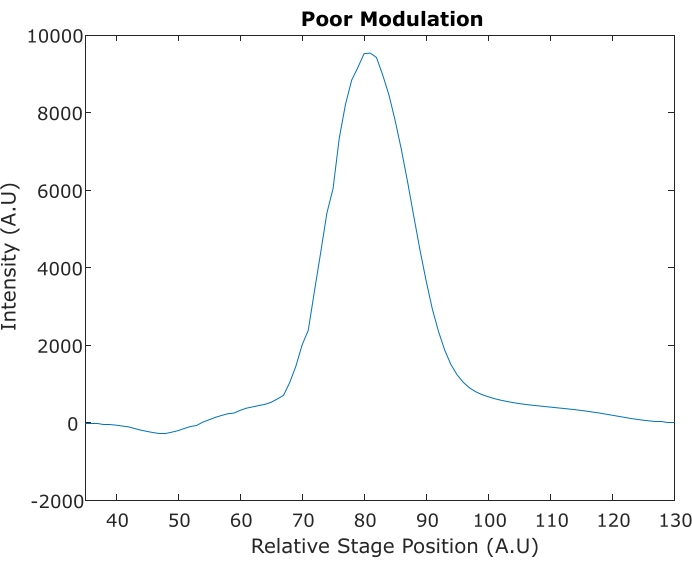
Figure 9: SRS spectrum resulting from a poor two-color SRS phase shift. A negative 2,994 cm-1 peak near stage position 48 is indicative of poor phase difference. Abbreviation: SRS = stimulated Raman scattering. Please click here to view a larger version of this figure.
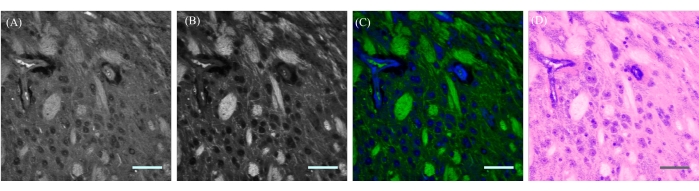
Figure 10: Generation of false two-color H&E staining of SRS images. (A) Raw protein SRS image from a mouse brain tissue at the 2,930 cm-1 transition. (B) Raw lipid SRS image from mouse brain tissue at the 2,850 cm-1 transition. (C) Merged and color-coded channels from A and B with lipid contribution in green and protein contribution in blue. (D) False H&E recoloring was performed on C to mimic H&E staining for pathological applications. Scale bar = 50 µm. Abbreviations: SRS = stimulated Raman scattering; H&E = hematoxylin and eosin. Please click here to view a larger version of this figure.
