Use of µTGGE to identify single nucleotide base changes in RNA editing events
Four edited genes were used for this protocol (Table 1), including the BFP gene produced in HEK293 cells (with C-to-U RNA editing by the deaminase enzymes of apolipoprotein B mRNA editing enzyme complex; APOBEC115), the EGFP gene containing the ochre stop codon (TAA) produced in HEK293 cells (with A-to-I RNA editing by adenosine deaminase acting on RNA 1; ADAR116), and the AT2G16586 and AT5G02670 nuclear genes in A. thaliana17,18 (with U-to-C RNA editing). Single nucleotide differences between the edited and corresponding nonedited samples were confirmed by DNA sequencing. Then a µTGGE analysis was performed to examine the differences between the melting curves of the samples (Figure 3).
For the C-to-U RNA editing type, the nonedited sample with the original C base showed a longer melting pattern at the strand end-melting point than the edited sample with the modified U base (Figure 3A). For the A-to-I(G) RNA editing type, the edited sample with the modified I(G) base displayed a longer melting pattern at the strand end-melting point than the nonedited sample with the original A base (Figure 3B). For the "reverse" U-to-C RNA editing type, two genes were analyzed. For the AT2G16586 gene, the edited sample with the modified C base showed a longer melting pattern between the strand initial-melting and strand end-melting points than the nonedited sample with the original U base (Figure 3C). However, a similar pattern was not observed for the other AT5G02670 gene (Figure 3D). Nonetheless, there was a distinct difference between the nonedited and edited types at the end-melting point. This shows that observing melting profiles clearly is an important step in distinguishing between nonedited and edited types.
Quantitative analysis of µTGGE melting patterns representing RNA editing events
Next, we calculated PaSS values11 to evaluate the reproducibility of the µTGGE-based melting profiles representing the four RNA editing events described above. The PaSS value provides a measure of how closely two melting patterns can be superposed, generating a higher value (maximum: 1) for highly similar melting patterns. Thus, PaSS values for comparisons of nonedited and edited samples are expected to be less than one. As shown in Figure 1B, the feature points of the melting patterns that corresponded to structural transitions from double-stranded to single-stranded DNA were used to calculate PaSS values. To eliminate experimental variables, computer-aided normalization was performed using two internal reference points: reference point #1, representing the position of the sample in double-stranded form (the leftmost lane), and reference point #2, representing the position of the sample in single-stranded form (the rightmost lane). The coordinates of the feature points were normalized to those of the internal reference points and were then used to calculate the PaSS values. Each experiment was repeated three times, and the average value was determined. As expected, the PaSS values of the four edited samples were lower than one (Figure 4). The PaSS values of the C-to-U and A-to-I RNA editing types were lower than those of the two U-to-C RNA editing types. This difference is likely related to the respective locations of the editing sites. Specifically, the C-to-U and A-to-I edited sites were located relatively close to the 5'-terminal ends (at positions 48 and 59 of the ~300 bp fragments, respectively), whereas the U-to-C edited sites were located near the middle of the fragments (at positions 152 and 169). These findings suggest that edited sites located at terminal positions can be detected using µTGGE more easily than those located toward the center of fragments.
Optimization of TGGE melting patterns to identify RNA editing events
As our previous results suggested that the PaSS value may vary depending on the specific position of the RNA editing site, we examined the differences between the melting patterns of a 300 bp fragment of the BFP gene (expressed in HEK293T cells) in which a C-to-U edited site was located close to the 5'-terminal end, close to the 3'-terminal end, or in the center of the fragment (Figure 5A). Prior to µTGGE analysis, the melting patterns of the nonedited fragment and three edited fragments were predicted using the uMelt HETS web-based tool. This analysis showed that the C-to-U modification would be expected to shift the melting curve to the left along the temperature axis (Figure 5B). The PaSS values calculated from µTGGE analyses of the nonedited and the three edited fragments were ordered as follows: 5'-terminal end RNA edit < 3'-terminal end RNA edit < center of 5'- and 3'-terminal ends RNA edit (Figure 5C). Notably, these PaSS values were consistent with the results predicted using uMelt. These findings indicate that nucleotide base differences located at the 5'- or 3'-terminal end result in larger variations between the PaSS values of edited and nonedited genes than nucleotide base differences located more centrally. In addition, these results suggest that prior knowledge of the differences between the melting profiles of edited and nonedited genes can be used as a guide to optimize gene fragments for RNA editing.
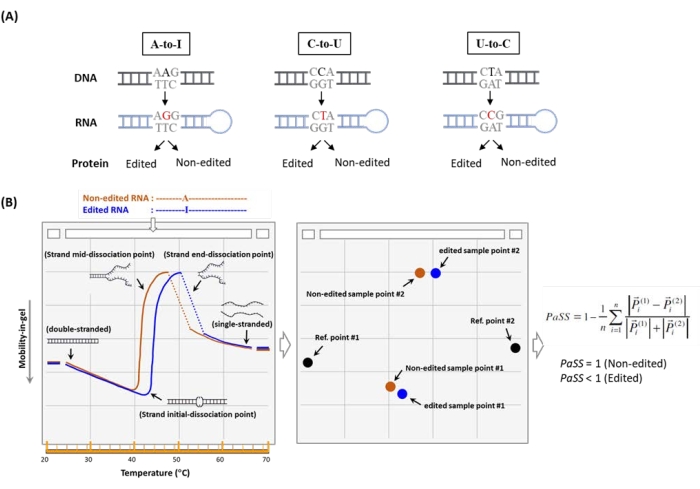
Figure 1: The procedure used to identify RNA editing by µTGGE. (A) The types of RNA editing events examined. (B) A schematic illustration of the typical melting profiles of edited and nonedited genes. In µTGGE, a sample migrates through a temperature gradient gel, producing a characteristic curvature. The feature points of the melting pattern are assigned and then processed to calculate a Pattern Similarity Score (PaSS) value. The PaSS calculation is performed as shown in the equation, where the vector P of each feature point is in its corresponding position and the function of temperature and mobility (i.e., vector P = P (T, m)). The superscripts (1) and (2) represent the edited and nonedited genes, respectively. Please click here to view a larger version of this figure.
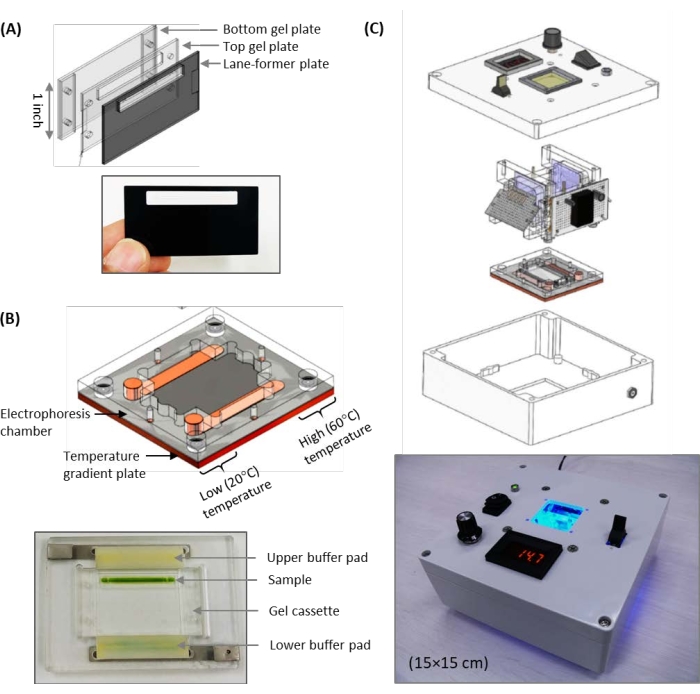
Figure 2: Illustrations and photographs of the palm-sized gel electrophoresis device. (A) Assembly of the three 1 in gel cassettes. (B) Illustration of the gel cassette holder with the temperature gradient plate. The photograph shows the positions of the upper and lower buffer pads, as well as that of the sample after initial loading. (C) Overview of the complete system, including the power supply, horizontal gel electrophoresis platform, and gel imaging system. Thissystem provides a viable solution for rapid, onsite polyacrylamide gel electrophoresis-based analyses. Please click here to view a larger version of this figure.
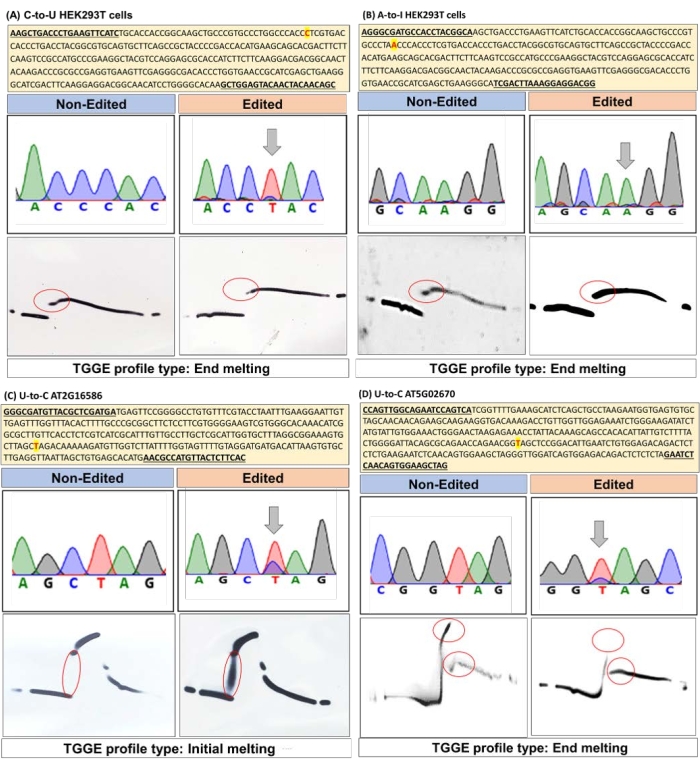
Figure 3: TGGE analyses of single nucleotide changes in RNA editing events. Melting profiles for three different RNA editing types were examined using four genes. (A) C-to-U RNA editing in BFP expressed in HEK293T cells. (B) A-to-I(G) RNA editing in EGFP expressed in HEK293T cells. (C) U-to-C RNA editing in the AT2G16586 gene from A. thaliana. (D) U-to-C RNA editing in the AT5G02670 gene from A. thaliana. The locations of the edited sites are highlighted in yellow (with red font), and the primer positions are underlined. The differences between the melting patterns of the edited and nonedited samples are indicated by red circles. Please click here to view a larger version of this figure.
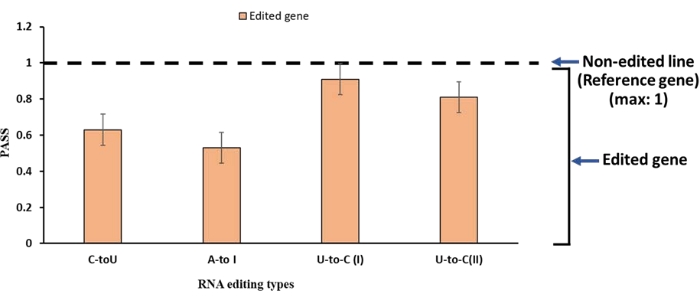
Figure 4: The average PaSS values for the four edited genes examined here. Error bars represent the standard deviation of three replicates. Please click here to view a larger version of this figure.
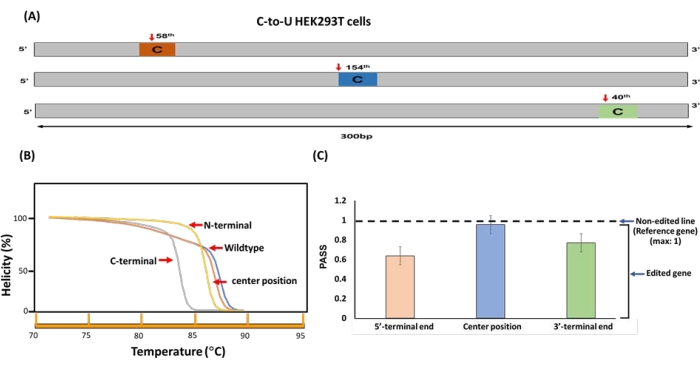
Figure 5: Position-specific PaSS analysis. (A) The C-to-U type RNA editing site in the BFP gene expressed in HEK293T cells was shifted to the 5'-terminal end, 3'-terminal end, or center of the gene fragment. (B) Theoretical prediction of the melting patterns of the nonedited and three edited fragments shown in (A). The predictions were performed using uMelt. (C) The average PaSSvalues of the edited genes shown in (A). Error bars represent the standard deviation of three replicates. Please click here to view a larger version of this figure.
| S.No | RNA editing | Source | Poste | Gene ID | Forward Primer | Reverse Primer | Sequence length | ||
| 1 | C-to-U | HEK293T cells | 48th | EGFP | AAGCTGACCC TGAAGTTCATC |
GCTGTTGTAGT TGTACTCCAGC |
324 | ||
| 2 | A-to-I | HEK293T cells | 59th | EGFP | AGGGCGATGC CACCTACGGCA |
CCGTCCTCCT TTAAGTCGA |
300 | ||
| 3 | U-to-C | Arabidopsis | 152th | AT2G16586 | GGGCGATGTT ACGCTCGATGA |
GTGAAGAGTAA CATGGCGTT |
301 | ||
| 4 | U-to-C | Arabidopsis | 169th | AT5G02670 | CCAGTTGGCAG AATCCAGTCA |
CTAGCTTCCAC TGTTGAGATTC |
300 | ||
Table 1: List of genes used in the current protocol
