1. Cleaning of the slide and coverslip for single-molecule experiments
- Before the assembly of the imaging chamber, clean and prepare both the coverslips and slides. Drill multiple pairs of holes on the glass slides using a drilling machine with diamond-coated drill bits (0.5-1 mm in diameter). If acrylic sheets are used, use a laser cutter to make precise holes (0.5 mm), as shown in Figure 1.
NOTE: Each pair of holes will act as an inlet and outlet for flow exchange for an individual microfluidic chamber. A representative CAD file for holes in an acrylic slide is available in Supplementary Coding File 1. Holes drilled on glass slides usually end up being 1.5x-2x larger than the drill bit size. - Clean the slides and coverslips with detergent. Rinse them thoroughly with deionized water. Place the slides and coverslips in a separate slide staining (Coplin) jar with water and sonicate in a bath sonicator for 30 min. For all the sonication steps, use a 70 W power setting.
- Remove the water and fill the Coplin jar containing the glass slides and coverslips with acetone to the brim (50-100 mL depending on the jar volume). In the case of acrylic sheets, use the same volume of premixed 50% (v/v) acetone solution or else the slides will become frosty. Shake the Coplin jar to ensure all the slides and coverslips are completely immersed in the solution and sonicate in a bath sonicator for 30 min.
- Remove the acetone and discard it in a waste container, pour methanol into the Coplin jars (50%, v/v for acrylic slides), and sonicate for 30 min. Both acetone and methanol are used to remove organic particles on the slides.
- Rinse the slides and coverslips with copious amounts of type 1 water and place them in a glass Coplin jar. Pour 1 M KOH solution into the jar and sonicate for 1 h. For the acrylic slides, use 0.5 M of KOH.
- Discard the KOH in an inorganic waste container. Rinse the jar with plenty of water and place the Coplin jars in a fume hood for piranha treatment. Do not perform piranha treatment for acrylic slides; directly proceed to step 1.9.
CAUTION: Piranha treatment should be done in a fume hood with proper gloves, safety glasses, and an apron for handling acids. Piranha solution is a strong oxidizing agent, and it removes any leftover organic particles from the slides and coverslips. - For preparing the piranha solution, gently pour a 2/3 volume of sulphuric acid (98%, v/v) into a glass beaker and fill the rest with hydrogen peroxide (30%, v/v). Mix the solution carefully with a glass rod, pour it into the Coplin jar, and leave the Coplin jar in the fume hood for at least 1 h.
- Decant the piranha solution from the Coplin jar in a discard container for acid waste kept inside the fume hood. Rinse the Coplin jar with a copious amount of type 1 water.
- Dry the slides and coverslips in a gentle stream of nitrogen gas and place them in a clean Coplin jar. The dried slides and coverslips are now ready for plasma treatment. Take a pair of slides and coverslips and place them in the plasma machine.
- Create a vacuum in the chamber. Turn the knob to the radio frequency of 8-12 MHz for generating plasma in the chamber. A purple glow inside the chamber will indicate the formation of a plasma in the chamber.
- Perform the plasma treatment for 8-10 min. Gently release the vacuum after turning off the plasma and proceed to step 2 for assembling the microfluidic imaging chamber.
2. Assembly of the microfluidic chamber
NOTE: The imaging chamber is created by sandwiching double-sided tape between a pre-cleaned coverslip and slide from the previous step as described below.
- Assemble the slides and coverslip after the plasma treatment as shown in Figure 1. Place the glass slide on a clean tissue paper. Cut the double-sided tape into ~2-3 mm wide strips and place them on each side of a pair of holes on the glass slide. Use a pipet tip to flatten out the tape or it will cause leakage from one channel to the other.
NOTE: Alternatively, cut the tape for the entire slide using a laser or automated plotter cutter (Figure 1). - Place the plasma-treated coverslip on the taped slide to close the chamber. Use a pipet tip to gently press the coverslip onto the taped regions to make the channel watertight.
- If using glass slides, seal off the edges using epoxy resin. Finally, each imaging chamber made from a slide and coverslip has a dimension of 10 mm x 2 mm x 0.1 mm (length x width x depth). Store the prepared microfluidic imaging chambers for 1-2 weeks in desiccated conditions (preferably between 10-25 °C).
NOTE: For the laser cut tapes used with acrylic slides, it is not necessary to use epoxy as the tape edges form a tight leakage-proof seal.
3. Making supported bilayers on glass substrate by vesicle fusion
NOTE: Crowded supported lipid bilayers are generated on the walls of the imaging chamber by the fusion of lipid vesicles prepared with doped PEG-lipids.
- Take a clean glass vial, preferably pre-cleaned using piranha solution. Add chloroform solution of 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC) and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000] (DOPE-PEG2000) at the desired mole fraction of PEG2000 lipids such that the final concentration after adding buffer will be 3 mM lipids in step 3.4. Prepare other membrane compositions of lipids similarly.
- Dry out the chloroform from the vial using a gentle stream of nitrogen with a small swirl so that the dried lipids are uniformly coated on the surface of the vial. Put the vial in a vacuum desiccator for 1 h. This will remove any residual chloroform in the glass vial. Add 1 mL of phosphate-buffered saline (PBS) and incubate overnight at 37 °C.
- Prepare small unilamellar vesicles (SUVs) as described below.
- Gently vortex for 1-2 min after overnight incubation until the solution turns turbid and milky.
- Take 100 µL of this solution in a small 500 µL centrifuge tube and sonicate for about 1 h in a bath sonicator (set at a 70 W power setting). The solution will become clear; if not, then sonicate for an additional 30 min. This step will generate SUVs of 50-100 nm in diameter.
NOTE: If preparing for the first time, characterize the SUVs using dynamic light scattering22,23 (DLS) or an equivalent method.
- Add calcium chloride (CaCl2, 3 M stock) to the sonicated vesicles such that its final concentration is 30 mM in the lipid solution.
- Cut a micropipette tip for injecting the sample solution such that it fits tightly in the hole. Mix the lipid solution and inject through one of the holes in the imaging chamber made in step 2. Wipe the excess solution that comes out of the chamber from the outlet with a clean tissue to prevent the contamination of adjacent channels.
- Leave this assembly in a humidified chamber for 90 min. Prepare a humidifying chamber by placing a wet tissue at the end of a 50 mL centrifuge tube. Place the slide sideways inside the tube with tube caps closed. The vesicles will fuse and generate a uniform bilayer on the glass surface.
- Wash the chamber thoroughly with a copious amount of PBS buffer (roughly 5 times the volume of the imaging chamber). Prevent the entry of air bubbles into the chamber while washing as this can generate defects in the membrane.
4. Microscope setup and single-particle imaging measurements
NOTE: Single-molecule experiments are carried out on an objective-based total internal reflection fluorescence24,25,26 (TIRF) microscope setup (Figure 2). TIRF imaging provides a better signal-to-noise ratio for single-molecule imaging, though epi-fluorescence microscopes can also be used under certain conditions (especially when the fluorescent biomolecules can be removed from the bulk solution by washing). The prism-type TIRF can be used but the objective-type is preferable for the ease of setting up microfluidics27. For objective-type TIRF, a high numerical aperture objective (100x magnification, usually commercially available as a TIRF objective) is recommended.
- Before conducting any experiment on mobility and assembly, align the TIRF microscope to ensure an optimal signal-to-noise ratio due to illumination by the evanescent field. During alignment, keep the laser power low, between 100 µW and 1 mW.
CAUTION: Laser safety glasses should be used during the aligning of the laser beam.- To align the microscope, prepare a fluorescent bead sample by adding them into the microfluidic channel at low concentrations (at ~100 pM) such that fluorescent spots from single beads do not overlap in the images.
- First, visualize the beads in epifluorescence mode. While illumination is in epifluorescence mode, move the M5 mirror translation stage (the mirror that reflects the laser to the dichroic) such that the beam coming out of the objective bends until it eventually is in a total internal reflection configuration.
- Verify whether TIR illumination has been achieved. When the TIR evanescent field is illuminating the bead sample, check that only the beads on the surface are visible and free-floating beads away from the surface are not observed.
- At the start of the experiment, set the laser power at the objective back focal plane to 5-10 mW to prevent photo-destruction (photobleaching) of the fluorophores.
- Keep the slide on the microscope stage and focus on the bare membrane (without any fluorophores) first. Usually, small traces of impurities in lipid membranes are sufficient to do this. Optional: Incorporate a small number of fluorescent beads or quantum dots (sub picomolar range) during step 3.1, such that only two to five fluorescent particles are present in the field of view to facilitate identifying the correct focus.
- Inject the sample (membrane-proteins, etc.) using a micropipette into the PEG-SLB-coated imaging chamber made in step 3.7.
- For measuring single-molecule binding kinetics, use a syringe pump to flow the labeled biomolecules through the inlet holes into the microfluidic chamber (Figure 3). Use a flow rate of 50-500 µL/min.
- Start movie acquisition before adding the solution into the imaging chamber. Acquire >5,000 frames at a frame rate of 25-50 frames/s under continuous flow until there is no further increase in the appearance of new spots on the membrane surface (Video 1). For the analysis of binding kinetics, follow the steps from 6.1.
- For single-particle tracking, add the labeled biomolecules at low concentrations (compared to the binding constant) to the channel using a micropipette. Optimize the concentration to a density of <0.1 particles/µm2 so that individual particles rarely cross paths (Video 2). This ensures the high fidelity of tracks recovered from the datasets. Incubate if needed (in case of poor membrane binding by biomolecules, ~10 min) in a humidifying environment and wash the chamber with the buffer.
NOTE: Washing is needed in case the binding is poor on the membrane and most of the fluorescent molecules remain in the solution. Otherwise, this may interfere with imaging and result in a poor signal-to-noise ratio. - For single-particle trajectory analysis, acquire 200-500 frames at 10-100 frames/s. Maintain the objective lens focused on the bilayer plane with minimal stage drift during the acquisition interval.
5. Image acquisition for counting subunits in a protein assembly
NOTE: Image acquisition for estimating stoichiometry requires continuous bleaching of fluorophores and detecting the number of steps until no more fluorophores are emitting fluorescence.
- For measuring subunits, add the relevant concentration of labeled biomolecules (example: see results; ClyA was added at 25 nM concentration) to the microfluidic imaging chamber and incubate (wash if necessary). Incubate the slide in a humidifying chamber at the desired temperature for the required amount of time (example: 37 °C and 60 min for the assembly of ClyA).
- Perform image acquisition for single-molecule photobleaching as described below.
- Wash the imaging chamber with a buffer before imaging (if necessary). Place the slide on the microscope and adjust the focus to visualize the labeled molecules.
- Set the laser power to a level at which the bleaching of the fluorophore occurs gradually (~1-5 min). Ideally, for each assembled biomolecule, keep the photobleaching step rate >10-20 imaging frames apart. Acquire the images until all the molecules are completely photobleached (Video 3).
NOTE: To determine the total duration of the photobleaching trajectory required, plot the average intensity of individual spots with the number of imaging frames and determine the time constant by fitting an exponential decay function. The total duration of the acquisition can be set to 5-10 times the time constant.
- Perform a time-dependent assembly measurement by starting movie acquisition as soon as the sample is introduced into the chamber and acquiring new photobleaching movies at fixed time intervals after membrane binding from different segments of the PEG-SLB.
6. Image and data analysis
- Perform single-particle binding and tracking as described below.
- Extract single-particle trajectories from the movies acquired above, as shown in Figure 4. For detecting single particles and tracking them, use the MATLAB package, u-track28, or the Trackmate29, a plugin in ImageJ, which also provides a robust single-particle tracking algorithm. Follow the steps for setting up u-track analysis as shown in Supplementary File 1.
NOTE: The critical parameters for single-particle tracking are the standard deviation of the particle size (in pixels) and the gap closing length. Use 1 and 0, respectively, for these parameters as the most stringent criteria for tracking. - To calculate the binding rate constants, first estimate the number of particles binding to the membrane surface over time. Use the MATLAB file binding_SPT (Supplementary Coding File 2) with the u-track output folder to identify and plot the number of particles appearing on the membrane obtained from step 6.1. Fit the observed kinetics to appropriate kinetic models (example: single exponential fit to determine the time constant, the inverse of which can be assigned to the apparent rate constant)30 to extract the rate constants (see results, Figure 5).
- The mean-squared displacement (MSD) of the diffusing particles (such as DNA tracer in PEG-SLBs, Figure 6) indicates the nature of diffusion. Use the MATLAB package MSD_ISD (Supplementary Coding File 2) with the u-track output folder to obtain the MSD plot as a function of lag time (Figure 6B).
- To identify heterogeneous diffusing species, calculate instantaneous squared displacement (ISD) distributions as shown in Figure 6C. ISD2, defined as (Xt+τ – Xt)2 + (Yt+τ− Yt)2, where lag time, τ = 2, is preferred as it reduces noise. Filter out short trajectories with less than three frames to reduce noise.
- Use ISD_analyzer (Supplementary Coding File 2) in MATLAB with the u-track output to plot the ISDs of the tracked particles (Figure 6C).
- Extract single-particle trajectories from the movies acquired above, as shown in Figure 4. For detecting single particles and tracking them, use the MATLAB package, u-track28, or the Trackmate29, a plugin in ImageJ, which also provides a robust single-particle tracking algorithm. Follow the steps for setting up u-track analysis as shown in Supplementary File 1.
- Perform single-molecule photobleaching analysis as described below.
- To estimate the stoichiometry of the membrane complexes, perform a photobleaching analysis on the time-dependent intensities of the fluorescent complexes (Video 3). For this, apply a drift correction algorithm (drift_correct_images, Supplementary Coding File 2) based on the autocorrelation of movie frames to extract the translation drift. This assumes that the assembled structures are largely immobile during image acquisition. Run the MATLAB package Extract_traces_1C (Supplementary Coding File 2) to detect particle intensity time traces from the movies.
- Use the MATLAB code Stepcount_immobile (Supplementary Coding File 2) to perform the step detection for the intensity time traces. In case the particles are not immobile, use the u-track package to extract the location of moving particles. This set of xy-coordinates can be mapped to the raw movies to extract the intensity values of the moving particle as it diffuses laterally on the membrane. Use the MATLAB package utrack_Int (Supplementary Coding File 2) with the output from u-track analysis for this option.
- Perform a correction for incomplete labeling of the biomolecules with fluorophore tags to estimate the correct protein complex stoichiometry. Determine the labeling efficiency with a UV-VIS spectrophotometer by measuring the absorption of dye and protein to estimate the concentration. Calculate the labeling efficiency as the molar ratio of the dye to the protein.
NOTE: Some dyes also absorb at wavelengths close to 280 nm, and, hence, this absorption contribution by the dye should be accounted for during the concentration calculation. - Perform a binomial correction to account for the incomplete labeling efficiency of the fluorophore in the following manner by using the MATLAB code Step_correction (Supplementary Coding File 2) with the data obtained from step counting in step 6.2.2. For example, for a pore-forming toxin like Cytolysin A, which forms a dodecameric ring-like structure, apply a binomial correction using the following formula:
nk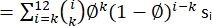
where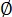 = labeling efficiency, n = number of photobleaching steps, and s = actual counts. Use the MATLAB routine Stepcount_immobile to recover the corrected oligomeric species. Convert the frequency of oligomers (si) to mass fractions (Figure 7C; Supplementary Coding File 2).
= labeling efficiency, n = number of photobleaching steps, and s = actual counts. Use the MATLAB routine Stepcount_immobile to recover the corrected oligomeric species. Convert the frequency of oligomers (si) to mass fractions (Figure 7C; Supplementary Coding File 2).
NOTE: A set of u-track analyzed files are included in Supplementary Coding File 3. The MATLAB codes in Supplementary Coding File 2 can be run with these data sets.
Monitoring the binding of ClyA protein on PEGylated membranes
After step 4.5, the binding kinetics are estimated by plotting the number of particles binding to the membrane surface over time (Video 1). As ClyA protein binds to a membrane with 5 mol% PEG2000 lipids,the particle density increases and reaches saturation (Figure 5). An exponential decay fit to the bound particles (cyan circles) gives the time constant (τb) for the membrane binding (notably, the initial time points [red circles] are not fit in this case).
Mobility of DNA tracer on crowded membranes
We commonly use DNA tracers (DNA anchored to the membrane with a tocopherol), lipophilic tracer dyes (e.g., DiI), or membrane proteins (e.g., ClyA) to characterize the membrane under PEG-mediated crowding. Lateral diffusion of labeled tracer molecules (25–100 pM) can be monitored by imaging the membrane upon particle binding. In the absence of any PEG polymer in the membrane, most tracers (especially those extending outside the bilayer) display restricted diffusion on SLBs (Figure 6A). With small levels of PEG2000 in the membrane (0.5–2 mol%), the bilayer lifts away from the underlying surface, and the tracer molecules can diffuse without constraints. On the other hand, extreme confinement is observed at a high concentration of PEG (20 mol%), where PEG molecules are in a dense brush regime, with a variety of behaviors displayed in between. MSD plots for these three conditions are shown in Figure 6B. Based on our characterization of POPC/DOPE-PEG2000 bilayer membranes, we recommend a 1–3 mol% DOPE-PEG2000 fraction that induces crowding in the mushroom regime. Above 7.5 mol% PEG2000-lipid, we observe the onset of the brush regime, and compositions until 25 mol% PEG2000-lipid can be employed to induce crowding and confinement. The intervening 4–7% PEG2000-lipid conditions corresponding to the transition between the two regimes are poorly characterized and should be avoided.
Assembly of ClyA on crowded membranes
The intensity trajectories of individual diffraction-limited spots of ClyA protein after incubation on the crowded membrane (Figure 7A,B) display a large number of distinct photobleaching steps, suggesting the formation of various assembly intermediates31. ClyA, being a transmembrane protein, interacts with the negatively charged glass surface in the absence of PEG and will assemble very poorly. At 7.5 mol% PEG2000 lipids, the assembled complexes were measured and are plotted in Figure 7C. After correcting for the labeling efficiency (0.9 in this case), the final distribution of oligomers displays the dodecameric ClyA species as the dominant structure, consistent with structural data32.
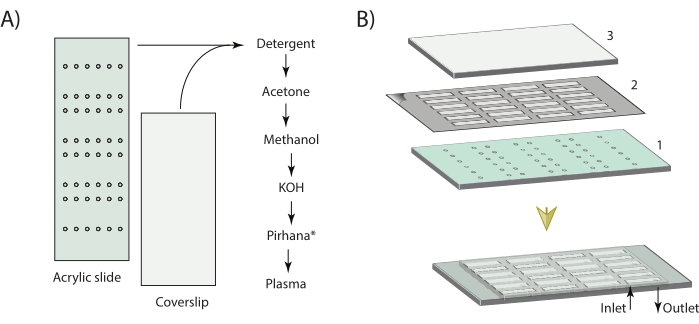
Figure 1: Schematic for the cleaning steps and microfluidic chamber assembly. (A) The glass slides and the coverslips are cleaned with sequential sonication in detergent, acetone, methanol, KOH, and piranha solution, with multiple rinsing by type 1 ultrapure water between each step. The slides and coverslips are then dried with nitrogen gas before plasma treatment. (B) The microfluidic imaging chamber is then assembled using a pre-cut double-sided adhesive tape that binds the slide to the coverslip. *Acrylic slides are not treated with piranha solution, and the acetone, methanol, and KOH are used at a 50% (v/v) concentration of that used for coverslip cleaning. Please click here to view a larger version of this figure.

Figure 2: TIRF microscope setup. A typical objective-type TIRF microscope setup adapted on an inverted microscope is shown with essential optics highlighted. M1–M5 are mirrors to steer the laser beam. Lenses L1 and L2 are used to expand the laser beam, and the L3 lens focuses the laser beam onto the back focal plane of the objective lens. I1 and I2 are the Iris diaphragms used to align the laser beam. A shutter is used to control the laser beam illumination, critical for preventing unnecessary photobleaching of the fluorescence molecules. Please click here to view a larger version of this figure.
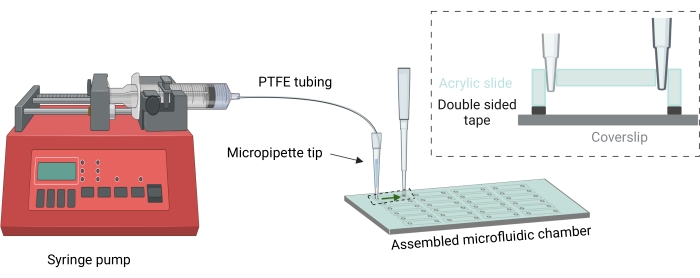
Figure 3: Membrane binding kinetics flow setup. For conducting the binding experiment, a thin PTFE tubing (0.022 in ID x 0.042 in OD) is used to flow the sample with the help of a syringe pump (image not to scale). The end of the tubing is connected to the imaging chamber outlet with the help of a micropipette tip. The discard is collected in a small microtip reservoir plugged into the outlet, and the volume introduced is always greater than 10 times the volume of the imaging chamber. Inset figure showing cross section of the imaging chamber. (image not to scale) Please click here to view a larger version of this figure.
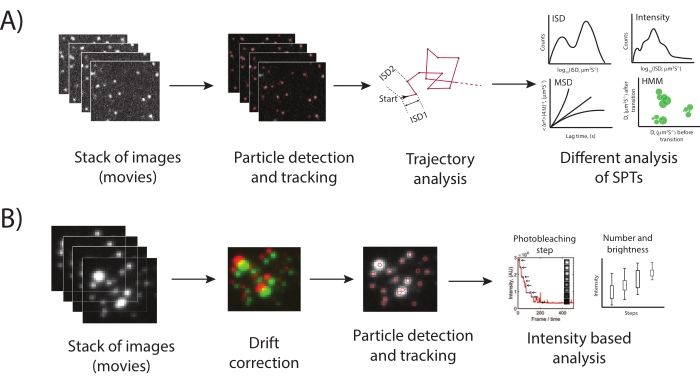
Figure 4: Pipeline for analysis from single-particle trajectories and photobleaching movies. (A) The acquired movies were analyzed using u-track in MATLAB to obtain the trajectories as (x, y) and the intensities (i) of each particle, frame-by-frame. These trajectories were then used to calculate instantaneous square displacement (ISD), ISD1, and ISD2. The ISDs can then be plotted as histograms to identify mobile species. Alternately, mean squared displacement (MSD) plots inform whether the motion is Gaussian, confined, or super-diffusive33,34. Hidden Markov Model35 (HMM) analysis can be employed to distinguish different diffusive states of a single particle. (B) For the photobleaching analysis, the movies were first corrected for drift in the system (if necessary), followed by particle detection or tracking using u-track. Apart from estimating the intensity distribution23 (and other particle features), the intensity trajectories can be analyzed for the number of photobleaching steps using step-finding algorithms36,37,38. Please click here to view a larger version of this figure.
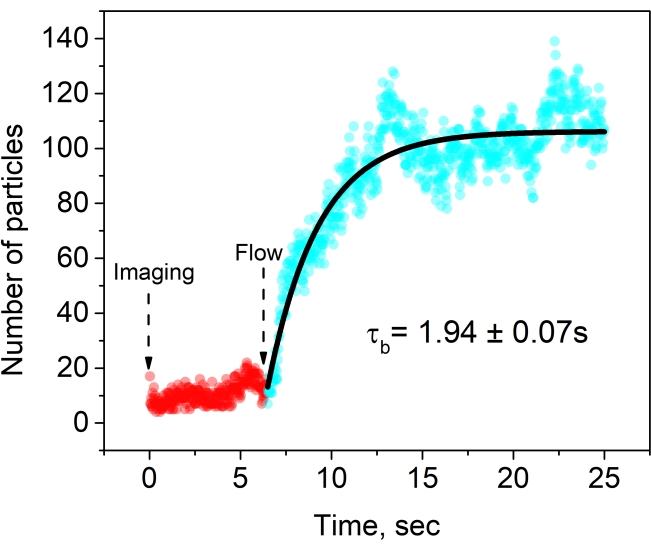
Figure 5: Binding of ClyA to supported lipid membranes with 5 mol% DOPE-PEG2000. A typical binding curve is obtained after counting the number of particles in each frame identified by u-track. Imaging is initiated before the flow of the membrane-binding ClyA protein. An exponential decay function (black line) fit to particle numbers (cyan circles) is used to recover the time constant for the binding of ClyA to the membrane. Please click here to view a larger version of this figure.
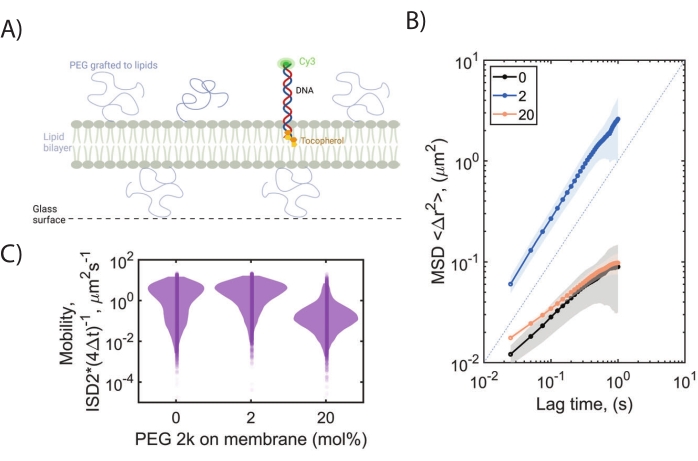
Figure 6: Mobility of lipophilic DNA on PEG-lipid bilayer membranes. (A) Schematic for the lipophilic DNA tracer in the POPC/DOPE-PEG2000 membrane is shown. (B) Mean squared displacements calculated from the single-particle tracking for different PEG-SLBs are shown. In 2 mol% DOPE-PEG2000, DNA tracer displays pure Brownian behavior compared to hindered mobility in the absence of the polymer (dashed line is included for reference). On the other hand, the same DNA tracer deviates away from pure Brownian diffusion, indicating sub-diffusive motion with reduced mobility in the presence of 20 mol% DOPE-PEG2000. (C) ISD2 distribution for the diffusion of lipophilic DNA tracer in two different PEG2000 lipid membranes is compared to bare SLBs. Please click here to view a larger version of this figure.
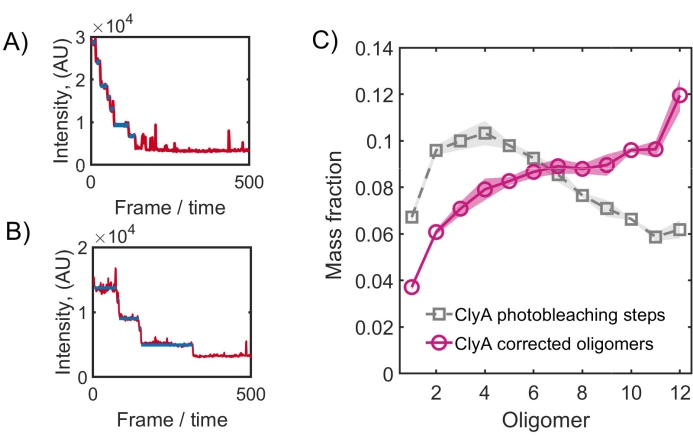
Figure 7: Photobleaching traces and the assembly of ClyA on lipid bilayer membranes. (A,B) Representative time traces show photobleaching steps of ClyA nanopore complex detected in step 6.2.1. for an assembled ClyA complex with 7 and 5 steps, respectively. (C) The photobleaching step distribution (grey) for ClyA (25 nM) incubated on 64.7% POPC: 27.8% Cholesterol: 7.5% DOPE-PEG2000 membrane for 60 min at 37 °C is shown. After correcting for the incomplete labeling efficiency, the estimated mass fraction of various oligomeric species (magenta) is shown. Please click here to view a larger version of this figure.
Video 1: Binding of ClyA on the membrane. Monitoring Cy3-labeled ClyA protein binding to the SLB membrane containing 5 mol% DOPE-PEG2000. The bound particles are detected (cyan circles) by u-track, and tracks are plotted for five subsequent positions in their trajectory. Please click here to download this Video.
Video 2: Diffusion of DNA tracer on the 2 mol% PEG membrane. A movie for Cy3-labeled DNA anchored to the lipid membrane via tocopherol in the 2 mol% PEG2000 membrane. Please click here to download this Video.
Video 3: Photobleaching movie. A movie of assembled oligomers of Cy3-labeled ClyA molecules on the membrane containing 7.5 mol% DOPE-PEG2000. Larger complexes are evident from the several fold higher intensities compared to a single protein, as well as from the multiple photobleaching steps when particle intensity is observed over time under continuous illumination. Please click here to download this Video.
Supplementary File 1: Information on how to use u-track and different MATLAB codes in step 6. Please click here to download this File.
Supplementary Coding File 1: A representative computer-aided design (CAD) file for making holes in the acrylic slide and cutting tape for the entire slide. Please click here to download this File.
Supplementary Coding Files 2: Compressed folder containing associated MATLAB codes needed for image and data analysis in step 6. Please click here to download this File.
Supplementary Coding Files 3: Sample data for image and data analysis in step 6. The MATLAB codes are also available at https://github.com/sgmaurya/SMTrack_Analysis. Please click here to download this File.
