Comprehensive validation of the analytical method was conducted according to SANTE/11312/2021 guidelines6, encompassing assessments of linearity, ME, recovery, and repeatability.
For the linearity assessment, matrix-matched calibration curves were constructed using spiked blank samples at multiple concentration levels (ranging from 5 to 600 µg/kg). The determination coefficients (R2) for most of the selected pesticides were found to be higher than or equal to 0.99, indicating a highly linear relationship between concentration and response. The lowest calibration level (LCL) of 5 µg/kg was chosen, adhering to the established maximum residue limit (MRL) established of 10 µg/kg for food monitoring purposes22.
To evaluate the ME, the slopes of the multiclass pesticides´ calibration curves were compared between pure solvent and matrix-matched calibration conditions. As an illustrative example, Figure 2 shows the comparison of the curves in the solvent and each of the three matrices for carbofuran. The ME was calculated using equation (1)7, yielding percentages that signify signal enhancement (positive percentages) or signal suppression (negative percentages).
Matrix effect (%) = 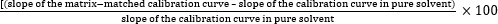 (1)
(1)
The presented ME classification system, based on percentage ranges, provides insights into the impact of the matrix on the pesticide signals, aiding in the interpretation of analytical findings. In all cases for carbofuran, a positive ME greater than 20% was obtained. However, the findings from the generation of matrix-matched calibration curves revealed a relatively consistent ME of less than 20% (classified as a soft ME) for most pesticide/variety combinations (see Table 2 and Figure 3).
To evaluate the accuracy and repeatability of the analysis, blank samples were spiked with pesticides at three different concentration levels (10, 100, and 400 µg/kg; n = 5 for each concentration). The results in Figure 4 demonstrate the count of pesticides whose average recovery percentages were within the acceptable range of 70-120% for each type of avocado. Furthermore, Table 3 presents detailed data for all the specific values obtained. A significant proportion of the tested pesticides exhibited recovery percentages falling within the specific range, with relative standard deviation (RSD) values below 20%.

Figure 1: Schematic representation of the QuEChERS method with ammonium formate employed for the extraction of pesticide residues from avocado samples. Abbreviations: QuEChERS = Quick-Easy-Cheap-Effective-Rugged-Safe; IS = internal standard; PSA = primary-secondary amine; GCB = graphitized carbon black; QC = quality control; GC-MS/MS = gas chromatography-tandem mass spectrometry. Please click here to view a larger version of this figure.
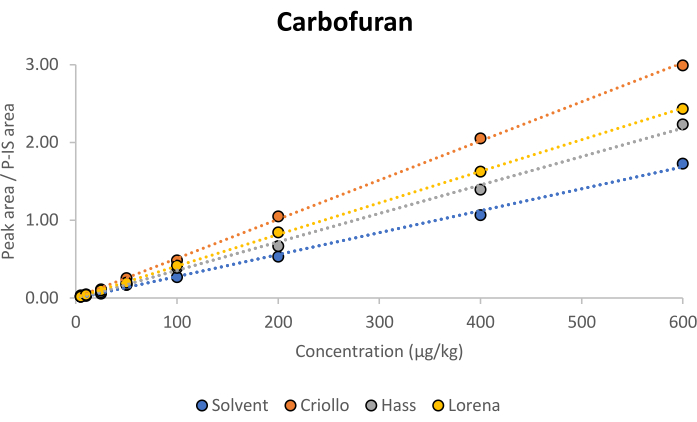
Figure 2: Comparison of the calibration curves in the solvent and matrices for carbofuran. Solvent: y = 0.0028x – 0.0054 and R2 = 0.9974; Criollo: y = 0.0050x + 0.0050, R2 = 0.9994, and ME = 80%; Hass: y = 0.0037x – 0.0109, R2 = 0.9977, and ME = 30%; Lorena: y = 0.0041x + 0.0053, R2 = 0.9998, and ME = 42%. Abbreviations: ME = matrix effect; P-IS = procedural internal standard. Please click here to view a larger version of this figure.
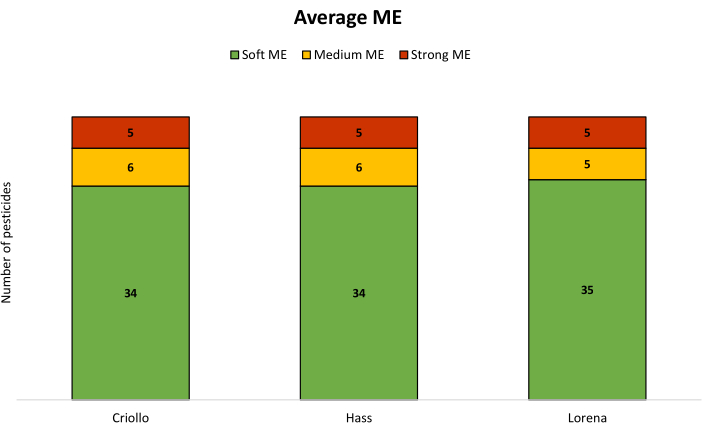
Figure 3: Number of selected pesticides categorized by their respective ranges of ME for avocado varieties. The classification of ME is based on three categories: soft (values between −20% and 20%), medium (values ranging between −20% and −50% or between 20% and 50%), and strong (values exceeding 50% or falling below −50%). Abbreviation: ME = matrix effect. Please click here to view a larger version of this figure.
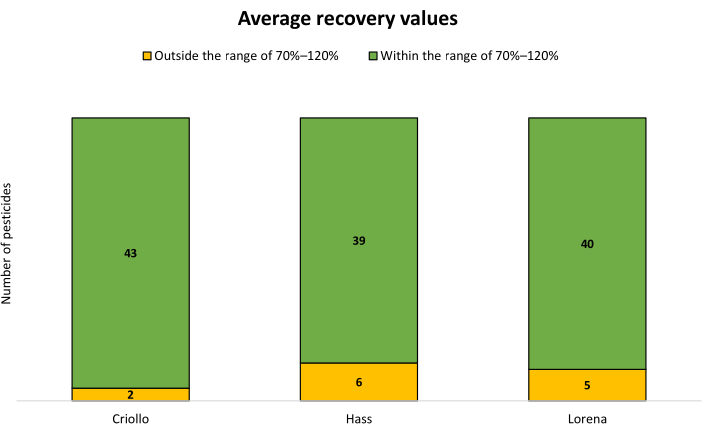
Figure 4: Number of pesticides that fall outside and within the acceptable recovery range spiked at 10, 100, and 400 µg/kg (n = 15) in the three avocado varieties. Please click here to view a larger version of this figure.
Table 1: Retention times, quantifier, and qualifier transitions utilized in GC-MS/MS analyses of the selected pesticides, along with the P-IS and I-IS. Abbreviations: P-IS = procedural internal standard; I-IS = injection internal standard; GC-MS/MS = gas chromatography-tandem mass spectrometry; HCB = hexachlorobenzene; α-HCH = alpha-hexachlorocyclohexane; β-HCH = beta-hexachlorocyclohexane; 4,4´-DDD = 4,4´-dichlorodiphenyldichloroethane; 4,4´-DDE = 4,4´-dichlorodiphenyldichloroethylene; 4,4´-DDT = 4,4´-dichlorodiphenyltrichloroethane; TPP = triphenyl phosphate; EPN = ethyl nitrophenyl phenylphosphonothioate. Please click here to download this Table.
Table 2: Matrix effect values (%) for the selected pesticides in different avocado varieties during the validation of the final analytical method. Abbreviations: HCB = hexachlorobenzene; α-HCH = alpha-hexachlorocyclohexane; β-HCH = beta-hexachlorocyclohexane; 4,4´-DDD = 4,4´-dichlorodiphenyldichloroethane; 4,4´-DDE = 4,4´-dichlorodiphenyldichloroethylene; 4,4´-DDT = 4,4´-dichlorodiphenyltrichloroethane; TPP = triphenyl phosphate; EPN = ethyl nitrophenyl phenylphosphonothioate. Please click here to download this Table.
Table 3: Recovery values and their corresponding RSDs in parentheses (n = 5 at each spiking level), both in %, for the selected pesticides in different avocado varieties during the validation of the final analytical method. Abbreviations: RSDs = relative standard deviations; HCB = hexachlorobenzene; α-HCH = alpha-hexachlorocyclohexane; β-HCH = beta-hexachlorocyclohexane; 4,4´-DDD = 4,4´-dichlorodiphenyldichloroethane; 4,4´-DDE = 4,4´-dichlorodiphenyldichloroethylene; 4,4´-DDT = 4,4´-dichlorodiphenyltrichloroethane; TPP = triphenyl phosphate; EPN = ethyl nitrophenyl phenylphosphonothioate. Please click here to download this Table.
Supplementary File 1: Mass spectrometric spectra of all pesticides. Abbreviations: HCB = hexachlorobenzene; α-HCH = alpha-hexachlorocyclohexane; β-HCH = beta-hexachlorocyclohexane; 4,4´-DDD = 4,4´-dichlorodiphenyldichloroethane; 4,4´-DDE = 4,4´-dichlorodiphenyldichloroethylene; 4,4´-DDT = 4,4´-dichlorodiphenyltrichloroethane; EPN = ethyl nitrophenyl phenylphosphonothioate. Please click here to download this File.
