Gタンパク質共役受容体(GPCR)は、すべての細胞表面タンパク質の中で最大かつ最も多様なファミリーの一つを構成する。脊椎動物、無脊椎動物、植物、酵母、及び粘菌においてだけでなく、原生動物や初期の二胚葉後生動物での存在は、GPCRは、シグナル伝達1にリンクされ、最も古い分子の一つであることを示しています。彼らの自然な活性化リガンドは、ペプチド、生体アミン、匂い物質、糖タンパク質、光子2を含む外部刺激の広い多様性を含んでいる。このように、これらの受容体 – リガンドのシグナル伝達系は、生理学的プロセスの多種多様に関与している。広いスペクトル機能性は、ヒトの疾患の広い範囲をカバーする治療薬の開発のためにそれらを最適です。現在の薬物標的の約50〜60%がGPCRは3,4で表される。製薬業界では彼らの偉大な重要性に加えて、GPCRは、開発のためのスポットライトにも記載されています一般的には種特異的な殺虫剤5,6及び農薬の新世代。多くのGPCRの天然リガンドはまだ同定されていないため、これらはオーファンGPCRに分類される。これらの受容体の脱オーファン化は生物で、その生理的役割の理解を改善し、新薬申請7のための推定標的を明らかにすることができる。
ゲノム時代以来、逆薬理学戦略は広く8のGPCRの脱オーファン化するために適用される。アプローチは、オーファン受容体は、生物学的抽出物から、または合成化合物のライブラリーから、その活性化リガンドへの「フック」「魚外」として使用されていることを意味します。目的のGPCRは、したがって、クローン化し、続いて細胞発現系でトランスフェクトされる。最も一般的に使用される方法では、受容体活性化は、二次メッセンジャー分子の濃度の変化を測定することによって決定される9 </suP>。主な受容体スクリーニングアッセイ( 例えば 、イクオリン)10や蛍光カルシウム指示薬( 例えば 、フルオ-4)11カルシウム感受性生物発光タンパク質に依存しています。受容体発現細胞前リガンドスクリーニングに蛍光カルシウム指示薬を負荷された蛍光ベースのアッセイは、それらが、それらの使いやすさ、短い読み取り時間、およびスクリーニングの柔軟性のハイスループットスクリーニングを可能にするという利点を有する単板12上に複数のオーファン受容体。
ここでは、蛍光ベースのカルシウム動員アッセイを十分に説明し、 キイロショウジョウバエの短いニューロペプチドF(sNPF)受容体の脱オーファン化過程で示されている。このneuropeptidergicシグナル伝達系は、もともとマーテンスらにより特徴付けられた。 2002年には、チャイニーズハムスター卵巣(CHO)細胞で行っカルシウム生物発光アッセイで1314と風水ら。、2003年のアフリカツメガエル卵母細胞に15を使用して電気生理学的ア ッセイとは。 sNPFシグナル伝達系の存在は、それが摂食の調節、増殖、ストレス反応、運動、および概日リズム16を含む広範囲のプロセスに関与している節足動物門の門に限定されると思われる。
昆虫におけるneuropeptidergicシグナル伝達システムの研究は、殺虫剤の開発のための新たな目標につながる可能性だけでなく、その機能についての知識はまた、多くのシグナル伝達系は、一般的によく進化17を通じて保存されている他の生物に向かって外挿することができる。過去10年間では、大きな進歩は、昆虫の神経ペプチドGPCRの脱オーファン化プロセスで行われている。これらの努力にもかかわらず、受容体の少数がその同族リガンドに整合されており、配列情報のための負荷新しいオーファンGPCRは、ゲノミクス18の急成長に利用できるようになりました。広く適用される技術9,18であることが証明された蛍光に基づくカルシウム動員アッセイのような中/高スループットスクリーニングアプローチが利用できるので、非常に貴重である。
蛍光に基づくカルシウム動員アッセイここに記載されるように、ヒト胚性腎臓293T(HEK293T)細胞株で行い、受容体活性化の際に細胞内カルシウム濃度の変化を測定するために蛍光プローブを使用する。受容体の高発現および翻訳レベルを保証するために、コザックコンセンサス配列19は、その後、発現ベクター(哺乳動物細胞株のために、例えば 、pcDNAベクター系)中にクローニングされた受容体コード配列の5 '末端に付加される。その配列情報に基づいて、オーファンGPCRの内因性Gプロテイン – 結合を予測することは困難であるように一人で、受容体の活性化の後に変調されたセカンドメッセンジャー分子( 例えば 、カルシウム又はcAMP)は、多くの場合、事前のリガンド同定には不明のまま。この問題を回避するために、ほとんどのGPCRはと相互に作用し、G qのファミリー ( 例えば 、マウス、Gα15または[ここで使用するヒトGα16)またはキメラGタンパク質( 例えば 、Gαのqi5)の無差別Gタンパク質は、カルシウムの放出を誘導することができる20,21,22共発現させることが。その受容体へのリガンドの結合の際に、GPCRは、特定の細胞内経路の活性化をもたらすコンフォメーション変化を受ける。 Gαサブユニット16に静止条件下で結合したグアノシン二リン酸(GDP)分子は、グアノシン三リン酸(GTP)分子によって置換される。これは、Gα及びGβγサブユニット16内ヘテロ三量体Gタンパク質の解離を引き起こす。 Gα16サブユニットはホスホリパーゼC&を活性化させる#946;今度はジアシルグリセロール(DAG)とイノシトール三リン酸(IP 3)、その結果、膜結合ホスファチジルイノシトールビスリ ン酸(PIP 2)を加水分解する(PLCβ)、。 IP 3は、細胞質全体に拡散し、細胞質へのカルシウムの放出を誘導する小胞体の膜に存在するIP 3依存性カルシウムチャネルを活性化するであろう。
受容体活性化によりカルシウム放出は、数秒以内に発生したFluo-4アセトキシメチル(AM)11のように、カルシウム感受性色素を用いてスクリーニングアッセイの前に細胞をロードすることによって検出することができる。 AMエステル基は、細胞膜を横断するフルオロフォアを可能にし、一旦細胞の内側の細胞質エステラーゼによって切断される。その結果、蛍光色素の負電荷が細胞から拡散し、カルシウムイオンと相互作用することを可能にするのを防ぎ、マスクされていない。蛍光信号OfはのFluo-4は、ナノモル範囲内カルシウム濃度を含有する静止条件下で細胞中にごくわずかである。カルシウムが受容体活性化の際に放出されたときしかし、信号は、本明細書大規模な信号対雑音比を確保し、100倍以上に濃度依存的に増加させることができる。のFluo-4はまた、それが適切な細胞の広範囲の生理学的に関連するカルシウムの変化を測定すること、345 nMでの K d(カルシウム)の周り[カルシウム]を報告するための大きなダイナミックレンジを示す。のFluo-4の励起は488nmで発生し、放出蛍光を525nmで11で測定される。蛍光イメージングプレートリーダー(FLIPR)23、カッセル、又はをFlexStation(基地局装置)12のような蛍光光度は、すべてのウェルについて同時に化合物の添加および受容体活性化の際のFluo-4シグナルの検出を可能にする媒体/ハイスループット系であるアッセイプレート中。ここに記載のカルシウム動員アッセイは、ステーションに依存していますデバイス96ウェルマイクロプレートシステム。
のSoftMax Proソフトウェア(ソフトウェア)は、データ分析のために、並びに局装置を操作するために使用される。プログラムはすぐに96ウェルフォーマットのグラフのような結果が表示されます。複数のウェルは、同じグラフ上に、これらのウェルの結果を比較するために同時に選択することができる。各列のウェルの相対蛍光単位(RFU)値は同時にウェルに化合物を添加する前に開始し、受容体活性化後の蛍光シグナルを測定した後に継続して、2分間の期間にわたって測定される。活性化化合物は、蛍光シグナルの急速な増加をもたらす、細胞に添加されるまで、典型的には、アゴニスト曲線の傾向は、ベースラインと整列する。ピーク高さは、ウェル中の最終アゴニスト濃度と相関している。ピークの後、蛍光シグナルは徐々にベースラインレベルに向かって降下する。 RFU測定のCAnは、リガンドのEC 50値(半最大有効濃度)を決定するために濃度-応答曲線に変換すること。一般に、少なくとも3つの独立したスクリーン、一連の濃度の3つの複製を含む各々は、信頼性の濃度 – 応答曲線を作成するために行われるべきである。
これは、実験計画のいくつかのポジティブ及びネガティブコントロールを含めることをお勧めします。まず、トランスフェクション制御は、既知のリガンドと受容体の実装、つまり 、テストする必要があります。これはトランスフェクション試薬が正常に動作したかどうかを検証することができます。細胞株および陰性対照( 例えば 、洗浄緩衝液)の内因性受容体に対するアゴニストでの対照実験の組み込みはまた、細胞の健康および生存率を監視し、洗浄緩衝液を用いて汚染された可能性を除外するために推奨されている自動fluoreを引き出す可能性が要因香り応答。頻繁に使用される作動薬は、アセチルコリン受容体を活性化するPAR 1選択的アゴニスト、又はカルバコール、として作用するプロテアーゼ活性化受容体-1(PAR 1)由来のペプチドである。空の発現ベクターでトランスフェクトされた細胞は、活性化合物は、細胞の内因性受容体と相互作用することを排除するために試験されるべきである。以下のプロトコールに記載されたいくつかのパラメータの最適化は、異なるシグナル伝達系に必要とされてもよい。完全な蛍光に基づくカルシウム動員アッセイの概略図を図1に示されている。
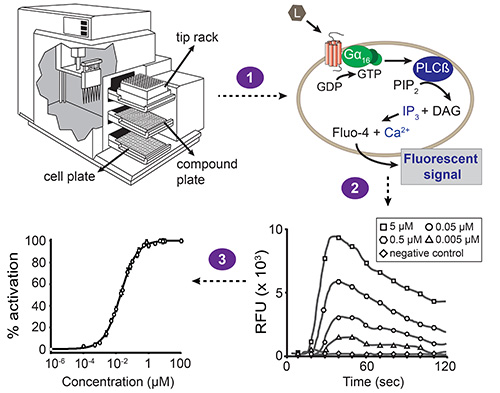
図1。蛍光ベースのカルシウム動員アッセイの全体的なスキーム。自動液体処理と同時蛍光測定は、駅で実施しているソフトウェアによって駆動されるデバイスマイクロプレートリーダー。セルプレートのための1、化合物のプレートとチップラック:局装置は3引き出しが含まれています。ビルドのピペッターを転送し、化合物プレートの1列からセルプレート(ステップ1)の対応する列への化合物。細胞プレートの各ウェルには、目的のGPCRと無差別Gα16サブユニットで同時トランスフェクトされたHEK293T細胞の単層を含んでいます。化合物が受容体を活性化すると、Gα16に結合したGDPは、GTPに置換されます。 Gα16サブユニットは、その後、Gβγ複合体から解離し、今度はホスファチジルイノシトールビスリ ン酸(PIP 2)ジアシルグリセロール(DAG)とイノシトール三リン酸(IP 3)を生じる加水分解するホスホリパーゼCβ(PLCβ)を活性化する。 IP 3は、カルシウムintの放出を誘導する、小胞体の膜に存在するIP 3依存性カルシウムチャネルを活性化する細胞質O。 (細胞は、化合物添加の前にロードされると)のFluo-4カルシウムの相互作用は、蛍光シグナル(ステップ2)をもたらす。ソフトウェアは、相対蛍光単位時間の関数で(RFU)値として結果を提示し、ピーク高さは、濃度依存的にリガンド濃度と相関する。これらのデータは、次いで、リガンド-受容体対(工程3)のEC 50値を決定するために、濃度-応答曲線に変換することができる。
