As proof of principle, gene expression was assessed to explore differentiation dynamics during cardiac development. In zebrafish, cardiac progenitors arise from a mesodermal population of cells that migrate to the anterior lateral plate mesoderm where they fuse to form the linear heart tube. Prior to fusion, cardiac progenitors begin to express the transcription factor nkx2.5 (NK2 homeobox 5), which is thought to be the earliest specific marker of cardiac progenitors16,17. Here, a previously described BAC transgenic fish Tg(nkx2.5:ZsYellow)18, abbreviated nkx2.5:ZsY was used to examine cardiac differentiation markers in single cells at the 18 somite stage, 18 hr post fertilization (hpf)12. This was the earliest time point at which ZsYellow signal was visually detectable. As previously described for this transgenic line, ZsYellow labels cardiac progenitors, as well as a few extra-cardiac cells that give rise to the pharyngeal arch endothelial cells at 28 hpf19 (Figure 1A-B, data not shown). At 18 hpf, nkx2.5:ZsY embryos were dissociated into a single cell suspension then stained with Sytox Blue to exclude dead cells. Live, ZsYellow positive, Sytox Blue negative cells were FACS sorted using either a MoFloXDP or Sony SH800Z sorter equipped with 100 µm nozzle (Figure 1C-F). To assess purity of the sorted population and post-sort viability, an aliquot of sorted cells were stained with Sytox Blue and evaluated the percentage of events that fell within the original sorting gate (Figure 1G-J).
After sorting, cells were entered into an integrated microfluidic circuit (IFC) chip work flow (Figure 2A) according to manufacturer's instructions. A hemocytometer, combined with trypan blue exclusion was used to measure cell diameter, concentration, and viability (Figure 2B, and data not shown). Cell buoyancy was optimized (6.5:3.5 cells:buffer) according to manufacturer's instruction, and cells were loaded onto an IFC to capture 5-10 µm diameter cells. To assess capture efficiency, fluorescence and/or bright field signals were imaged at all capture sites, a tiling function in FIJI was used to stitch a single picture of the microfluidics plate. Each capture site contained 0, 1, or >1 individual cells (Figure 2C-F). As expected from FACS enrichment, captured cells expressed ZsYellow (Figure 2G). Capture efficiency exceeded 90% in 5 individual experiments using nkx2.5:ZsY sorted cells, and at least 70% of capture sites were occupied by a single cell (data not shown). Cells were lysed, RNA isolated, cDNA synthesized and specific target genes were amplified, all according to manufacturer's instructions.
A subset of cell capture sites were selected for qRT-PCR analysis as described in the protocol above. Specifically, elongation factor 1a (efl1a)20 and glyceraldehyde-3 phosphate (gapdh)21 were assayed as housekeeping genes expected to be expressed in every cell; GATA binding protein 4 (gata4)22 and NK2 homeobox 5 (nkx2.5) as early cardiac progenitor markers; ISL LIM homeobox 1 box 1 (isl1) as a second heart field marker23,24; and myosin light chain (myl7) and ventricular myosin heavy chain (vmhc) 25 to mark ventricular cardiomyocytes. Gene-specific probes were validated using cDNA from 48 hpf embryos (Figure 3). qRT-PCR was used to assess relative gene expression of these genes in 45 single cells from 18 hpf embryos, a no-template negative control, and a pooled population positive control containing cDNA from all 96 capture sites. Raw CT values for 40 representative cells and controls are shown in Table 1. Notably, from the pool of 46 cells examined, 6 cells were excluded from analysis because all gene expression CT values exceeded CT=35.0, and it is unknown whether these high CT values are attributable to true, very low expression levels or sample degradation. Since the range of CT values for the housekeeping gene, ef1a, was too broad to compare gene expression between samples, and many CT values exceeded 30, CT values were visualized as a heat map (Figure 3). Comparing across samples, substantial heterogeneity in gene expression was observed, and cells were classified cells as Type 1-5 based on expression pattern (Figure 3).
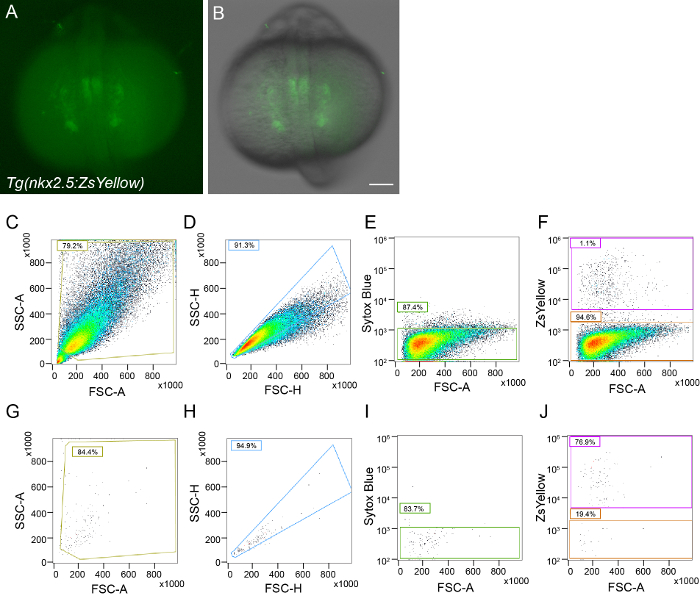
Figure 1. Single cell isolation of zebrafish nkx2.5:ZsY positive cells at 18 hpf. Whole mount images of representative Tg(nkx2.5:ZsYellow) embryo at 18 hpf with (A) ZsYellow fluorescence alone or (B) merged with bright field image. (C-F) Representative FACS gating strategy to enrich for ZsYellow positive cells and (G-J) post-sort analysis with (C,G) FSC/SSC size gating, (D,H) doublet discrimination, (E,I) Live/Dead gating, and (F,J) sorted population. Scale bar is 100 µm. Please click here to view a larger version of this figure.
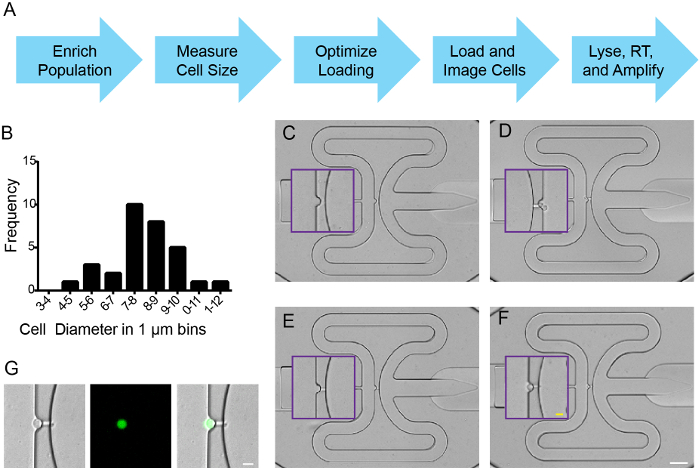
Figure 2. Single cell capture of zebrafish nkx2.5:ZsY positive cells. (A) Work flow. (B) Cell size distribution for sorted cells from Tg(nkx2.5:ZsYellow) embryos isolated at 18 hpf. (C-F) Representative cell capture events on IFC plate where (C) is an empty well, (D) contains two cells, (E) has a single cell lodged in the fluidics channel as a "channel capture", and (F) is a single captured cell. Purple boxes mark inset for magnified view of capture sites. (G) Single cell capture with brightfield, ZsYellow and merged images. (C-F) White scale bar is 50 µm; yellow scale bar is 10 µm. (G) Scale bar is 10 µm. Please click here to view a larger version of this figure.
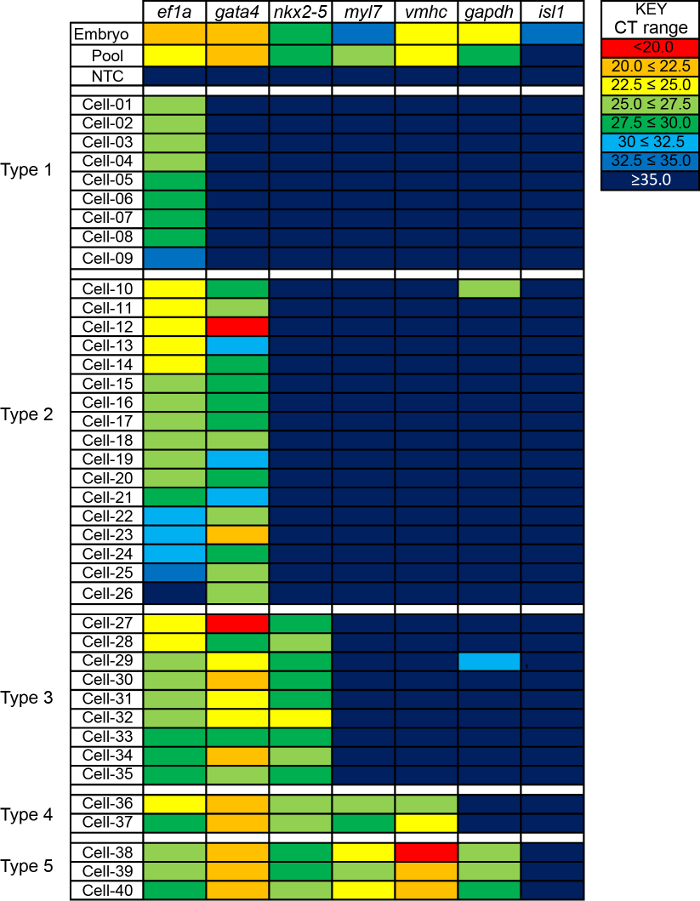
Figure 3. Gene expression analysis of capture single zebrafish nkx2.5:ZsY positive cells. Gene expression by qRT-PCR in positive controls (embryos at 2 days-post-fertilization, pooled cDNA from single cells), negative control (no-template) and 40 single cells (Cell 01-40). Raw CT values were color coded based as described in the Key. Ef1a = elongation factor 1a, gata4 = GATA binding protein 4, nkx2.5 = NK2 homeobox 5, myl7 = myosin light chain 7, vmhc = ventricular myosin heavy chain, gapdh = glyceraldehyde-3 phosphate, and isl1 = ISL LIM homeobox 1. Please click here to view a larger version of this figure.
| Cycle Threshold (CT) | ||||||||
| ef1a | gata4 | nkx25 | myl7 | vmhc | gapdh | isl1 | ||
| Embryo | 20.69 | 20.92 | 28.59 | 32.87 | 26.30 | 27.00 | 33.57 | |
| Pool | 26.4 | 22.4 | 27.7 | 27.4 | 24.5 | 29.2 | 40.0 | |
| NTC | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Type 1 | Cell-01 | 25.1 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 |
| Cell-02 | 25.4 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-03 | 26.3 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-04 | 27.4 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-05 | 28.0 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-06 | 28.2 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-07 | 29.3 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-08 | 30.7 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-09 | 34.3 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Type 2 | Cell-10 | 23.5 | 28.0 | 40.0 | 40.0 | 40.0 | 26.3 | 40.0 |
| Cell-11 | 23.5 | 27.2 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-12 | 23.7 | 17.9 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-13 | 24.1 | 32.4 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-14 | 24.9 | 27.5 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-15 | 25.9 | 28.6 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-16 | 26.0 | 28.1 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-17 | 27.0 | 27.7 | 40.0 | 40.0 | 40.0 | 38.5 | 40.0 | |
| Cell-18 | 27.0 | 27.4 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-19 | 27.0 | 30.2 | 40.0 | 40.0 | 40.0 | 38.2 | 40.0 | |
| Cell-20 | 27.0 | 28.2 | 40.0 | 40.0 | 40.0 | 39.2 | 40.0 | |
| Cell-21 | 27.7 | 30.3 | 40.0 | 40.0 | 40.0 | 38.6 | 40.0 | |
| Cell-22 | 30.4 | 26.2 | 40.0 | 40.0 | 40.0 | 38.3 | 40.0 | |
| Cell-23 | 31.3 | 22.4 | 40.0 | 40.0 | 40.0 | 39.7 | 40.0 | |
| Cell-24 | 31.5 | 28.8 | 40.0 | 40.0 | 40.0 | 39.5 | 40.0 | |
| Cell-25 | 33.8 | 27.4 | 40.0 | 40.0 | 40.0 | 37.3 | 40.0 | |
| Cell-26 | 40.0 | 27.4 | 40.0 | 40.0 | 40.0 | 36.6 | 40.0 | |
| Type 3 | Cell-27 | 24.7 | 19.9 | 28.8 | 40.0 | 40.0 | 40.0 | 40.0 |
| Cell-28 | 24.8 | 28.4 | 26.3 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-29 | 25.1 | 23.5 | 27.6 | 40.0 | 40.0 | 32.5 | 40.0 | |
| Cell-30 | 26.3 | 21.4 | 27.5 | 40.0 | 40.0 | 39.9 | 40.0 | |
| Cell-31 | 26.9 | 24.8 | 28.2 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-32 | 27.0 | 22.5 | 23.8 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-33 | 27.8 | 27.6 | 27.7 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-34 | 28.1 | 21.9 | 26.6 | 40.0 | 40.0 | 37.7 | 40.0 | |
| Cell-35 | 29.3 | 26.5 | 28.1 | 40.0 | 40.0 | 38.3 | 40.0 | |
| Type 4 | Cell-36 | 24.8 | 20.3 | 26.0 | 27.4 | 26.6 | 40.0 | 40.0 |
| Cell-37 | 28.7 | 22.3 | 25.9 | 28.9 | 22.9 | 38.5 | 40.0 | |
| Type 5 | Cell-38 | 25.5 | 20.0 | 29.3 | 23.0 | 19.9 | 25.9 | 40.0 |
| Cell-39 | 25.9 | 21.8 | 28.6 | 25.1 | 21.7 | 25.7 | 40.0 | |
| Cell-40 | 28.9 | 22.0 | 27.0 | 23.0 | 21.3 | 27.6 | 40.0 | |
Table 1. Raw CT values.
