Advanced Imaging of Lung Homing Human Lymphocytes in an Experimental In Vivo Model of Allergic Inflammation Based on Light-sheet Microscopy
Summary
The here introduced protocol allows characterization of the lung homing capacity of primary human lymphocytes under in vivo inflammatory conditions. Pulmonary infiltration of adoptively transferred human immune cells in a mouse model of allergic inflammation can be imaged and quantified by light-sheet fluorescence microscopy of chemically cleared lung tissue.
Abstract
Overwhelming tissue accumulation of highly activated immune cells represents a hallmark of various chronic inflammatory diseases and emerged as an attractive therapeutic target in the clinical management of affected patients. In order to further optimize strategies aiming at therapeutic regulation of pathologically imbalanced tissue infiltration of pro-inflammatory immune cells, it will be of particular importance to achieve improved insights into disease- and organ-specific homing properties of peripheral lymphocytes. The here described experimental protocol allows to monitor lung accumulation of fluorescently labeled and adoptively transferred human lymphocytes in the context of papain-induced pulmonary inflammation. In contrast to standard in vitro assays frequently used for the analysis of immune cell migration and chemotaxis, the now introduced in vivo setting takes into account lung-specific aspects of tissue organization and the influence of the complex inflammatory scenario taking place in the living murine organism. Moreover, three-dimensional cross-sectional light-sheet fluorescence microscopic imaging does not only provide quantitative data on infiltrating immune cells, but also depicts the pattern of immune cell localization within the inflamed lung. Overall, we are able to introduce an innovative technique of high value for immunological research in the field of chronic inflammatory lung diseases, which can be easily applied by following the provided step-by-step protocol.
Introduction
Classic inflammatory disorders of the lung, such as allergic asthma and chronic obstructive pulmonary disease (COPD), are well known to be driven by an increased recruitment of activated lymphocytes into the pulmonary tissue1,2. Lymphocyte-released cytokines (e.g., IL-4, IL-5, IL-9, IL-13, IFN-γ and TNF-α) further promote chemotaxis of innate and adaptive immune cells, induce fibrotic airway remodeling or directly damage the lung parenchyma2. So far, the underlying mechanisms responsible for the pathological accumulation of lymphocytes within lung tissue are not yet fully understood. In analogy to tissue-selective T cell imprinting described for gut and skin homing, pulmonary dendritic cells (DCs) are obviously able to prime peripheral T cells for preferential lung infiltration, at least partly via the induction of CCR4 expression on the surface of lymphocytes3. Besides CCR4, airway-infiltrating T cells are also characterized by a particularly increased expression of the chemokine receptors CCR5 and CXCR3 compared to T cells within the peripheral blood1,4,5. Overall, existing data are consistent with the concept that lung homing of T lymphocytes under physiological or inflammatory conditions involves a number of different chemokine receptors and their respective ligands and thus crucially depends on a closely controlled collaboration between innate and adaptive immune cells1. Especially, during the initial phase of pathogen or allergen exposure, cells of the innate immune system respond to TLR stimulation or to IgE-mediated cross-linking by the immediate release of different chemoattractants, like LTB4, CCL1, CCL17, CCL22, CCL20, CXCL10 and PGD21,6,7. As a prime example, the interaction between PGD2 and the chemoattractant receptor CRTh2 is known to be of particular importance for chemotaxis of Th2 cells and thus appeared as promising therapeutic target in the clinical management of asthma. Indeed, patients with moderate asthma showed an improvement of symptoms and a significant increase of the forced expiratory volume in one second (FEV1) after treatment with a selective CRTh2 antagonist compared to the placebo group8,9. In a more progressed state of the inflammatory response, already recruited T cells are able to further amplify pulmonary lymphocyte accumulation via the release of IL-4 and IL-13 as potent stimuli for pulmonary DCs. Subsequently, these myeloid-derived innate cells up-regulate the expression of CCL17 and CCL22 in a STAT6-dependent manner1,10,11. Although the complexity of the described scenario still hinders a complete understanding of T cell lung homing, it offers a plethora of molecular targets for a potentially optimized therapeutic control of inflammatory or allergic pulmonary diseases. Therefore, there is an urgent need of innovative experimental techniques, which are able to further deepen and complement our knowledge in the field of T cell chemotaxis and lung homing.
Due to the fact that lung homing of lymphocytes within the human body is influenced by multiple cellular, humoral and physical parameters1, most of the existing experimental methods are not able to model the whole complexity of this immunological process. Instead, many standard protocols for the analysis of lung homing selectively focus on a specific aspect involved in the cascade of lymphocyte attraction, adhesion, migration and retention. Besides a purely descriptive determination of the mRNA or protein expression pattern of integrins and chemokine receptors on peripheral or lung-infiltrating lymphocytes and the complementary measurement of respective chemokine levels in blood, bronchoalveolar lavage (BAL) or pulmonary tissue12,13,14,15, well-established in vitro cell culture assays allow a functional characterization of lymphocyte adhesion or chemotaxis upon defined experimental conditions16,17,18. In principle, static in vitro adhesion assays monitor the binding capacity of cultured lymphocytes to an endothelial monolayer or to glass slides coated with recombinant endothelial adhesion molecules (e.g., MAdCAM-1, VCAM-1), while standard in vitro chemotaxis assays are usually applied in order to quantify the ability of lymphocytes to migrate along a chemokine gradient in a transwell system19. Both in vitro settings enable a controlled adjustment and modulation of experimental conditions, but on the other hand lack important variables known to critically impact on in vivo chemotaxis and adhesion of lymphocytes. Predominantly, static cell culture assays disregard the influence of shear forces caused by the permanent blood flow19 and potentially neglect the involvement of the surrounding immunological milieu and interacting non-lymphocyte immune cells, both present in a living organism. In order to overcome these limitations, the interpretation of results acquired in static in vitro chemotaxis or adherence assays need further validation in dynamic adhesion experiments under flow conditions20,21 and in in vivo models of inflammatory organ pathology19. Indeed, important conclusions on the regulation of T cell lung homing under inflammatory or allergic conditions could be drawn from animal studies analyzing genetically modified mice in defined models of different pulmonary diseases3,22,23. The quantitative comparison of lung infiltrating lymphocytes between wildtype mice and mice with a deficiency for a specific gene of interest represents a well-established and broadly used tool for defining the impact of particular cellular pathways or receptors on the disease-driven pattern of T cell distribution. However, in contrast to before discussed in vitro cell culture assays, a study design based on classical animal models lacks the ability to analyze and monitor primary human T cells directly derived from the blood or BAL of patients suffering from an inflammatory lung disease. Thus, it still remains challenging to functionally validate whether a diagnostically specified lung disease is able to imprint human lymphocytes for preferential lung tropism and how far clinical parameters might impact on this scenario. Recently, a very elegant in vivo approach was introduced in the context of inflammatory bowel diseases (IBD), which was able to overcome most of these limitations and opened new avenues for advanced translational studies on intestinal lymphocyte homing24. Taking advantage of protocols for solvent-based tissue clearing followed by cross-sectional light-sheet fluorescence microscopy as a powerful imaging tool, it was possible to visualize the infiltration and distribution of adoptively transferred human T cells in the intestine of colitic immunodeficient mice24. In particular, this experimental setting implemented two main innovations: (1) Primary human immune cells can be analyzed under experimentally defined in vivo conditions; (2) a rather large area of the diseased organ (about 1.5 cm x 1.5 cm) can be imaged in high resolution quality, followed by 3D-reconstruction. Moreover, several recent studies successfully established the use of solvent-based tissue clearing and light-sheet fluorescence microscopy as important tools for advanced lung imaging25,26. In order to benefit from this technological progress in the field of pulmonary immunology, we now adopted the system for analysis of lung homing.
The here presented protocol provides a step-by-step introduction how to purify and fluorescently label primary human T cells for transfer into mice with induced pulmonary inflammation and, moreover, describes in detail the subsequent process of light-sheet fluorescence microscopic imaging, including organ preparation and image processing. Overall, we hope to support future translational studies in the field of inflammatory or allergic lung diseases by introducing a sophisticated, but nevertheless feasible, experimental model for monitoring human lymphocyte lung homing upon in vivo conditions.
Protocol
Experiments involving animals were performed in accordance with protocols approved by the relevant local authorities in Erlangen (Regierung von Unterfranken, Würzburg, Germany). Mice were housed under specific pathogen-free conditions. The collection of human blood was approved by the local ethical committee and the institutional review board of the University of Erlangen-Nuremberg. Each patient gave written informed consent.
1. Induce Allergic Lung Inflammation in Mice
NOTE: As described in earlier studies27, the following experimental procedure allows inducing allergic airway inflammation in mice and, accordingly, triggers the accumulation of innate and adaptive immune cells in the BAL. The described protocol has been established in C57BL/6J mice, but adoption to other standard inbred strains should be possible.
See Figure 1A for a summary of the in vivo experimental procedure.
- Anesthetize mice by intraperitoneal injection of ketamine/xylazine.
NOTE: Only use C57BL/6J mice older than 6 weeks and with a body weight of at least 16 g.- Prepare ketamine/xylazine solution in PBS (12 mg/mL ketamine; 1.6 mg/mL xylazine) and determine the exact body weight of mice.
- Inject the first mouse with 8 µL/g bodyweight of ketamine/xylazine solution intraperitoneally and confirm deep anesthesia via the absence of the paw pinch reflex before proceeding to step 1.3.
- Freshly prepare 5 mg/mL of papain in PBS. Slowly pipette 10 µL (50 µg) of the papain solution into the nostril. Carefully monitor the mouse until awakening.
- Repeat steps 1.1 and 1.2 for all mice included in the experiment. Perform steps 1.1–1.3 on three consecutive days.
2. Purify and Fluorescently label Human Peripheral Blood CD4+ T Cells
NOTE: Process cells under sterile conditions.
- Collect 18 mL of full human blood from a peripheral vein. Perform Ficoll-Hypaque gradient centrifugation in order to select the fraction of peripheral blood mononuclear cells (PBMC).
NOTE: Collection and analysis of primary human material need to be approved by the local ethical authorities. Only a person with an adequate medical qualification is allowed to collect blood from a peripheral vein.- Collect blood in ethylenediaminetetraacetic acid (EDTA)-containing monovettes (1.6 mg EDTA/mL blood) in order to avoid coagulation.
OPTIONAL: Use buffy coat blood, a by-product of blood donation enriched in leukocytes, instead of complete venous blood. - Transfer blood into a conical 50 mL tube and dilute 1:2 in phosphate buffered saline (PBS). Carefully add 10 mL of Ficoll-medium (density 1.077 g/mL) as a bottom layer under the diluted blood. Centrifuge the sample (800 x g for 15 min at room temperature without brake).
- Carefully remove the tube from the centrifuge and transfer the PBMC-containing interphase into a new conical 50 mL tube. Discard the upper and bottom layer. Fill the PBMC-containing tube with PBS and centrifuge (300 x g for 10 min at 4 °C). Remove the supernatant.
NOTE: Optionally, if necessary, perform lysis of remaining erythrocytes. Carefully add 3 mL of ammonium-chloride-potassium (ACP) lysis buffer (155 mM ammonium chloride; 19 mM potassium hydrogen carbonate and 0.68 mM EDTA; pH 7.27) to the resuspended cell pellet and vortex the sample. Shake the tubes for 3 min. To remove the ACP lysis buffer, add 40 mL of PBS and centrifuge (300 x g for 10 min at 4 °C). Discard supernatant.
- Collect blood in ethylenediaminetetraacetic acid (EDTA)-containing monovettes (1.6 mg EDTA/mL blood) in order to avoid coagulation.
- Purify CD4+ T cells via CD4 microbeads.
NOTE: In general, there are several distributors of magnetic microbeads for purification of human CD4+ cells, which should result in comparable levels of cell purity. Product choice should be based on individual preferences. The following steps of the protocol (2.2.2–2.2.7) are adjusted to a defined CD4 microbead product and respective separation columns as further indicated in the Table of Materials. Some modifications of the protocol might be required in case that an alternative CD4 microbead product is selected.- Resuspend the PBMC pellet in a minimum of 80 µL PBS containing 0.5% fetal bovine serum (FBS) and 2 mM EDTA (PBS/FBS/EDTA-buffer) per 107 PBMC. Use a starting population of about 30 x 106 PBMC in order to end-up with approximately 3 x 106 to 6 x 106 purified human CD4+ T cells.
- Add 20 µL of CD4 microbeads per 107 PBMC, mix gently and incubate for 20 min at 4 °C. Fill the tube with PBS/FBS/EDTA-buffer (cooled down to 4 °C) and centrifuge (300 x g for 10 min at 4 °C). Remove supernatant.
- Place a separation column into a magnetic field. Rinse the column with 3 mL of PBS/FBS/EDTA-buffer. Resuspend the cell pellet in 500 µL of PBS/FBS/EDTA-buffer (cooled down to 4 °C) and transfer the cell suspension onto the rinsed separation column placed within a magnetic field.
- Rinse the separation column three times with 3 mL of PBS/FBS/EDTA-buffer (cooled down to 4 °C) and discard the effluent.
- Remove the separation column from the magnetic field and place it into a 15 mL tube. Elute the CD4+ cell fraction in 5 mL of PBS/FBS/EDTA-buffer by using the plunger.
- Fill the tube with PBS and centrifuge (300 x g for 10 min at 4 °C). Remove supernatant. Repeat this washing step twice.
- Determine the number of yielded cells by a standard method of choice (e.g., Neubauer chamber).
- Label CD4+ T cells with a fluorescent cell proliferation dye.
- Resuspend CD4+ T cells in PBS (up to 107 cells/mL; do not use less than 500 µL of PBS).
- Prepare a 6 µM solution of a red light-excitable cell proliferation dye (see Table of Materials) in PBS (pre-warmed to room temperature). Mix this solution 1:2 with the before prepared cell suspension and vortex thoroughly (3 µM final concentration).
- Incubate for 10 min at 37 °C (protected from light). Afterwards, add five volumes of RPMI medium containing 10% FBS and incubate for 5 min on ice (protected from light).
- Wash labeled cells three times in RPMI medium containing 10% FBS (centrifuge at 300 x g for 10 min at 4 °C). Resuspend labeled cells in PBS, store on ice and protect from light until use.
NOTE: Prolonged storage of cells (>60 min) might negatively impact on cell survival.
OPTIONAL: Determine the purity of the CD4+ cell fraction and the efficacy of the labeling procedure by flow cytometry. Resuspend 0.5 x 106 to 1 x 106 CD4+ cells in 100 µL of buffer (1% FBS, 2 mM EDTA in PBS) and add an anti-human CD4 antibody coupled with a fluorescent dye of choice. Incubate for 30 min at 4 °C. Add 1 mL buffer and centrifuge (300 x g for 10 min at 4 °C). Discard supernatant. Proceed with flow cytometric measurement of the sample (representative result is depicted in Figure 1B,C).
3. Adoptively Transfer Human CD4+ T Cells into Papain-exposed Recipient Mmice
NOTE: See Figure 1A for a summary of the in vivo performed experimental procedure. Perform cell transfer one day after the last intranasal administration of papain.
- Adjust the suspension of fluorescently labeled human CD4+ T cells (step 2.3.4) to a concentration of 1 x 107 cells/mL in PBS. Fill 100 µL of the cell suspension into a 1 mL/30 G-syringe.
- Carefully place the first papain-exposed C57BL/6J mouse (steps 1.1–1.3) into a restraint device for tail vein injection. Use the prepared syringe (step 3.1) to puncture the tail vein and slowly inject 100 µL of the cell suspension containing 1 x 106 labeled human CD4+ cells. Do not immediately pull out the needle after injection, but wait for additional 5 seconds in order to prevent discharge of the cell suspension.
- Release the mouse from the restraint device and proceed with the next animal. Keep at least one papain-exposed animal, which does not undergo transfer with fluorescently labeled cells to serve as negative control for light-sheet fluorescence microscopy.
4. Prepare Lung tissue for Light-sheet Fluorescence Microscopy
NOTE: The following steps are performed 3 h after transfer of fluorescently labeled human cells (step 3.2). The described experimental procedures including harvesting of lung tissue, fixation and solvent-based tissue clearing were adapted from currently described protocols24,28.
- Sacrifice the mouse by carbon dioxide (C02) inhalation.
- Perfuse the lung in situ with PBS containing 5 mM EDTA.
- Open the thorax via cutting along the sternum to expose the heart. Punctate the right ventricle with a 21 G cannula connected to a catheter.
- Open the left ventricle by cutting and thereby allow the blood and perfusion fluid to exit. Slowly perfuse the lung with 20 mL of ice-cold PBS containing 5 mM EDTA via the catheter.
- In order to perform in situ fixation of the lung, do not remove the catheter from the right ventricle. Slowly perfuse the lung with 2 mL of ice-cold 4% paraformaldehyde (PFA) solved in PBS. Remove the catheter and the cannula.
NOTE: PFA-based in situ fixation of lung tissue represents a well-established technique in order to prepare murine lung tissue for subsequent microscopic analysis29,30.
CAUTION: PFA is toxic and has to be handled under a hood with care. - Fill the lung with 0.75% agarose in situ.
- Prepare 0.75% agarose in PBS and keep it solid at 50 °C in a thermo-shaker until use. Remove the salivary glands of the mouse, cut the sternohyoid muscle and expose the trachea by sliding a forceps underneath.
- Punctuate the exposed trachea with a 30 G needle and replace it by a blunt 30 G catheter to prevent further damage of the trachea resulting in undesired leakage of agarose. Seal the junction between the inserted catheter and the trachea using a needle holder.
OPTIONAL: Combine the here described procedure with BAL collection to analyze infiltrated human cells in BAL as recently described in detail31 (see Figure 2E for representative results). - Carefully fill the airways with 0.75% agarose (cooled down to body temperature) via the catheter until complete unfolding of the lung; wait until complete solidification of the agarose. Remove the catheter and carefully harvest the lung in a darkened 2 mL tube filled with 4% PFA.
- For additional fixation incubate the sample in 4% PFA solved in PBS for 2 h at 4 °C under continuous rotation (31 rpm).
- Dehydrate tissue by subsequently incubating the sample under continuous rotation (31 rpm) at 4 °C in 50% ethanol (pH 9), 70% ethanol (pH 9) and 100% ethanol; each step for at least 4 h. At the end, perform a second incubation in fresh 100% ethanol for 4 h.
- Perform solvent-based clearing of the tissue using ethyl cinnamate (ECi). Therefore, transfer the sample into ECi. Incubate overnight at room temperature (under constant rotation) until the tissue appears translucent (see Figure 1D for representative images).
NOTE: Samples stored in ECi at room temperature (protected from light) are stable over several weeks to months.
5. Perform Light-sheet Fluorescence Microscopy of Wwhole Murine Lung Lobes
NOTE: Please see the Table of Materials for details on the light-sheet fluorescence microscope and the corresponding software, on which the following steps are based. Comparable systems by other manufacturers, however, can be used as well with developer-specific modifications of the following protocol. Before starting, get familiar with the microscope-specific operating manual and follow the technical instructions by the responsible person on site.
- Set up the light-sheet microscope.
- Turn on the light-sheet microscope and open the corresponding imaging software on the computer. Fill the sample chamber with filtered ECi (100 µm filter) and place it in its position between the laser beams.
- Use a small drop of organic solvent-stable glue to stick the lung to the sample holder. To keep the required penetration depth of the light-sheets as small as possible, set the lung lobe upright. Next, place the sample holder in its chamber.
- Set the refractive index of the objective to 3.5 when using ECi as clearing reagent.
- Adjust settings at the light-sheet microscope to detect human cells in the context of the whole organ.
- Select required lasers (filter for measurement > activate checkboxes of required lasers) and choose appropriate intensities in the control software (laser transmission control > use slider to set laser intensity > apply). Use an excitation wavelength of 488 nm (525/50 filter) to detect autofluorescent lung tissue and 640 nm (680/30 filter) to excite human cells labeled with a fluorophore emitting in the red spectrum.
- Focus on the sample excited at 488 nm and adapt the focus via the ‘chromatic correction’ tool at the excitation wavelength of 640 nm (use the slider to set chromatic correction > apply).
- Choose an appropriate zoom factor; use a low magnification overview (e.g., 6.3x) to determine the overall distribution of labeled cells within the lung tissue and magnified images (e.g., 32x) for detailed localization.
- Select a sheet width uniformly elucidating the whole organ or a specific section of interest (optics > use slider to set sheet width; usually between 20–40%). Define the sheet numerical aperture (NA) with higher NA creating sharper images (optics > use slider to set the sheet NA; for a murine lung lobe expanded to its physiological size a NA of 0.025% can often be used).
- Select the number of light-sheets to be used under ‘advanced measurement settings’. In case of bidirectional illumination, merge left and right light-sheets to create a homogenously illuminated image (advanced > merge lightsheets > select blend mode > use sliders to define overlap between both light-sheets).
NOTE: It is recommended to take advantage of the bidirectional illumination of the sample with three light-sheets each.
OPTIONAL: To further increase image quality, use the dynamic focus operation. This allows to move the focus in x-direction during image acquisition. - Define start and end positions of the z-stack to be measured and set the step size to 5 µm (xyz-table Z > scan range).
NOTE: Start and end positions of a z-stack depend on size and positioning of the lung lobe within the sample chamber. However, about 300 to 800 z-stacks are usually acquired for a single lung lobe (5 µm step size). - Save files using ‘autosave settings’ and start measurement to capture images. After finishing data acquisition, clean the ECi-contaminated sample holder and chamber under running water.
NOTE: Use the same settings to image several lung lobes that are supposed to be compared afterwards.
6. Post-image Processing and Quantification of Lung-accumulated Human Cells
NOTE: Please see Table of Materials for details on the post-imaging software for 3D analysis, on which the following steps are based. However, alternative post-imaging softwares can be used as well.
- Load the first image of a z-stack (activate the surpass button, file > open > select image) in order to initiate opening of all other images of the chosen z-stack and their automatic 3D reconstruction.
- To ensure correct display of x, y and z dimensions, check voxel sizes and adjust them if necessary (edit > image properties > enter voxel size manually). For the voxel size of z, enter the chosen step-size between two images (here 5 µm).
- Display each channel in a distinct color (edit > show display adjustment). Click on each channel one after the other to define a color of choice (in Figure 2A,B,C,D the autofluorescence signal is displayed in grey and labeled human CD4+ cells in red).
- Adjust intensity, black level and contrast for each channel (edit > show display adjustment) using the triangles in the channel bars or entering exact numbers in the channel settings. For intensity adjustment, use the right triangle. For black level adjustment, use the left triangle and for contrast adjustment, use the middle triangle.
NOTE: Use the lung derived from the control mouse without cell transfer (step 3.3) to differentiate between specific human cell-derived signals and unspecific background. - To quantify labeled cells within the pulmonary tissue use ‘add new spots’ in the toolbar and select ‘skip automatic creation, edit manually’ for manual cell counting.
NOTE: Alternatively, use the automatic cell counting tool under ‘spots’. However, manual counting allows to differentiate between specific and unspecific signals more precisely.- To compare the accumulation of human cells across different samples, define a lung cube with a specific volume using the 3D cutting tool (edit > crop 3D) (here 813 μm x 813 μm x 1,000 μm).
- To count accumulated cells select the 640 nm channel and define a ‘radius scale’ of 10 µm. Then, click on ‘edit’, check ‘select’ in the pointer menu and mark each cell with a dot via shift-click with the left mouse button. Find the overall number of counted cells displayed in the statistics.
NOTE: It is highly recommended to count several cubes per lung lobe (here five) to ensure reliability of results and compensate for scientist-dependent selection of the counted lung volume (quantification strategy and representative results are depicted in Figure 2D).
- Capture an image using the ‘snapshot’ tool and/or take a video using the ‘animation’ tool. Save adjusted files as .ims files (file > export).
NOTE: For representative purposes, show or hide the frame by checking or unchecking the frame in the toolbar. In addition, display images either as maximal intensity projection (MIP) (toolbar > volume > mode > check MIP) or use the ‘surface mode’ (toolbar > add new surfaces). The ‘surface mode’ has to be activated separately for each channel to highlight tissue architecture and/or cell bodies (representative images are shown in Figure 2C).
Representative Results
The presented protocol describes an experimental mouse model, which allows monitoring and quantifying the accumulation of adoptively transferred human T lymphocytes in the lung via light-sheet fluorescence microscopy. Figure 1A provides a schematic overview of the in vivo steps of the experimental schedule. In order to guarantee reliable results, it is of substantial importance to ensure a good quality of the isolated and fluorescently labeled human CD4+ T cells, which will afterwards be transferred into mice. As representatively depicted in Figure 1B,C, the above described procedure for microbead-based cell enrichment and subsequent fluorescence-labeling usually results in a flow cytometrically determined CD4+ T cell purity >95% and successful fluorescence-labeling of all CD4+ T cells. The quality and penetration depth of light-sheet fluorescence microscopy critically depends on an appropriate grade of tissue clearing. As demonstrated in Figure 1D, the here applied protocol for ECi-based tissue clearing was able to guarantee a high level of organ transparency, indicating a successful refractive index matching. Finally, a representative overall result of the described experimental protocol in form of fully processed light-sheet fluorescence microscopy images is demonstrated in Figure 2B,C,D and in the Supplementary Video. The autofluorescent signal (displayed in grey) provides a helpful tool for imaging the anatomic structure of the lung. The red signal represents lung accumulated human CD4+ T cells. Quantification of light-sheet fluorescence microscopy imaging allows determining the overall number of lung accumulated human CD4+ T cells per defined area of inflamed lung tissue. A strategy for quantification of human cell infiltration is illustrated in Figure 2E. The (optional) flow cytometric detection of fluorescently labeled cells within the BAL of recipient mice, as depicted in Figure 2F, can be used as a supplemental technique in order to confirm the successful tissue migration of adoptively transferred CD4+ T cells. Moreover, the preference of transferred human T cells for selective accumulation in inflamed lung tissue in the here described experimental setting was further supported by the fact that human CD4+ T cells could not be retrieved in the intestinal mucosa of recipient animals as depicted in Figure 2B (right panel).
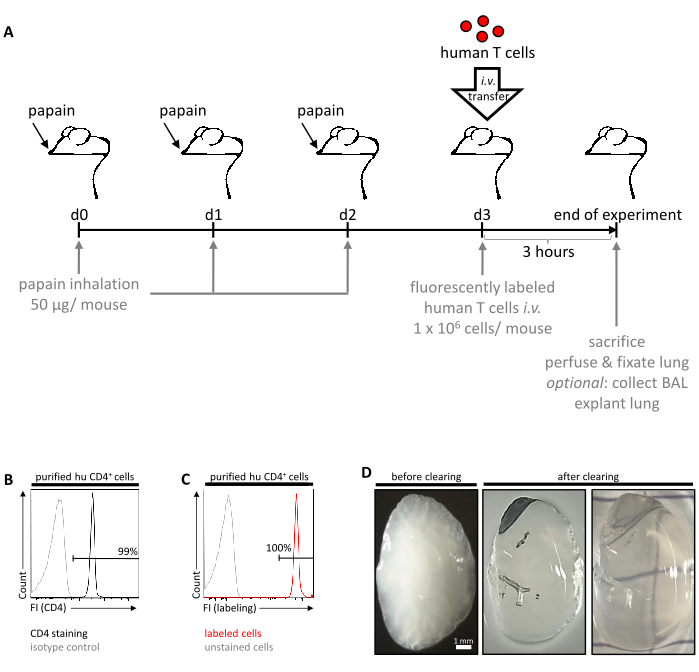
Figure 1: Schematic overview of the in vivo experimental workflow. (A) Allergic lung inflammation was induced in C57BL/6J mice by inhalation of papain (50 µg/mouse) on three consecutive days (d0, d1 and d2). The next day (d3), freshly isolated and fluorescently labeled human CD4+ T cells were adoptively transferred into mice via tail vein injection. After another three hours, mice were sacrificed, followed by in situ perfusion and fixation of the lungs. Finally, lungs were explanted and further analyzed ex vivo by light-sheet fluorescence microscopy. Optionally, BAL can be collected before lung explantation to perform extended ex vivo analyses of successfully extravasated and migrated human cells. (B) Representative histogram confirming the purity of the isolated CD4+ T cell fraction as determined by flow cytometry. Cells stained by an anti-human CD4 antibody or isotype control are displayed in black and grey, respectively. (C) Representative histogram confirming the efficacy of the fluorescence-labeling of human CD4+ cells prior to adoptive transfer. (D) Representative images of a perfused murine lung lobe before and after clearing with ECi. FI, fluorescence intensity. Please click here to view a larger version of this figure.
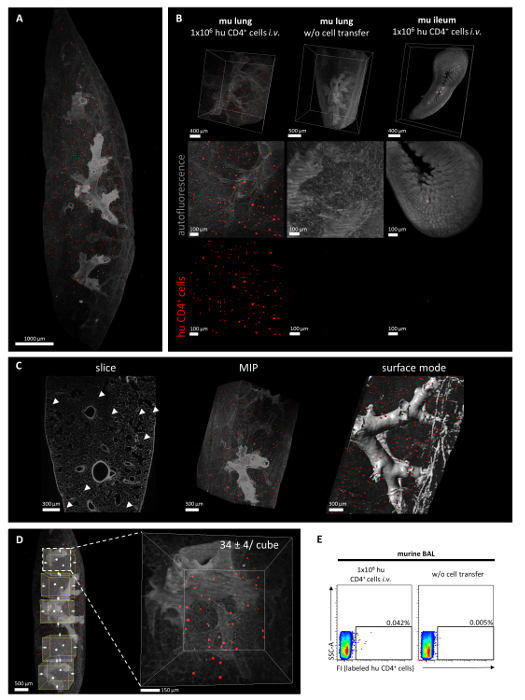
Figure 2: Representative analysis of the pulmonary accumulation of human CD4+ cells in the context of lung inflammation in vivo. Human CD4+ cells were isolated from peripheral blood using density gradient centrifugation followed by a magnetic microbead-based purification step. Human CD4+ cells were labeled using a red light-excitable cell proliferation dye and injected intravenously into papain-exposed C57BL/6J mice. After three hours, mice were sacrificed to collect and further analyze the lung tissue via light-sheet fluorescence microscopy. (A) Representative overview of a 3D reconstruction of murine, papain-exposed lung tissue (grey) three hours after intravenous injection of labeled human CD4+ cells (red) as recorded by light-sheet fluorescence microscopy. (B) Lung tissue of a recipient mouse three hours after cell transfer shown as overview or high magnification segment (left panel). Retrieved human CD4+ cells could be visualized as red signals and were either depicted in overlay with the autofluorescence signal of lung tissue or as single-channel image. In order to confirm the specificity of the detected signal, control lung tissue without intravenous cell transfer served as negative control (middle panel). In contrast to lung tissue, no labeled human CD4+ cells (red) could be detected in the ileum of the same recipient mouse (right panel). (C) Post image processing allows analysis of single slices of lung tissue as well as generation of 3D reconstructions of the whole organ segment that can be either represented as maximal intensity projection (MIP) or in surface mode. Representative images are depicted. (D) Quantification strategy is illustrated exemplarily in the same lung as already depicted in Figure 2A. Defined cubes of the analyzed lung segment were selected and the number of human CD4+ cells (red) was quantified for each cube. Results of a subsequently performed quantification (events/813 µm x 813 µm x 1,000 µm cube) are indicated as mean ± SEM. (E) As an additional option, flow cytometric detection of fluorescently labeled human T cells in the BAL of recipient animals can be performed in order to confirm the successful tissue migration of accumulated human CD4+ T cells. Please click here to view a larger version of this figure.
Supplementary Video: Representative video sequence showing accumulated human CD4+ cells within the murine lung tissue. 3D reconstruction of the murine lung displaying accumulated human CD4+ cells (red) within the context of the pulmonary tissue (grey) three hours after intravenous injection into the tail vein. Images are shown as MIP in the beginning and in the surface mode in the end. Images were acquired via light-sheet fluorescence microscopy and processed by post-imaging software for 3D analysis. Please click here to view this video. (Right-click to download.)
Discussion
The here described experimental setting provides the opportunity to monitor the lung homing capacity of primary human immune cells under in vivo inflammatory conditions and thereby relevantly complements classically performed in vitro adhesion and chemotaxis assays. To take into account specific anatomic organ characteristics of the lung, important aspects of immune cell homing (including chemotaxis and cell distribution within the target organ) as well as the clinical relevance and transferability of acquired data, we took advantage of three technical key features: (1) the analysis of primary human cells within a living murine organism; (2) the papain-mediated induction of pulmonary inflammation; and (3) light-sheet fluorescence microscopy of cleared lung tissue.
Although functional studies of adoptively transferred human immune cells within a living murine organism might be limited in some aspects due to potentially relevant incompatibilities in receptor-ligand-interactions between mice and men, the general concept of humanized mouse models has been established successfully as an important experimental tool in the field of translational medicine during the last decades24,32,33,34,35. Compared to classical animal models, studies in humanized mice offer the advantage to directly analyze patient-derived immune cells in an in vivo scenario and thereby to identify functional alterations imprinted by the particular disease32,36,37. Regarding the relevance of potential mismatches between defined receptors on the surface of human immune cells and their respective murine ligands or vice versa, former studies already indicated that human CD4+ T cells are able to interact efficiently with the murine adhesion molecules MAdCAM-1 and VCAM-1, which represent crucial ligands for integrin-mediated lymphocyte homing32. Accordingly, migration of intravascularly transferred human immune cells into murine gut tissue could successfully be blocked in vivo by treatment with the clinically used α4β7 integrin antibody vedolizumab32. However, even in case that a specific receptor-ligand-interaction of relevance might show a complete human/murine mismatch, it will presumably be possible to overcome this limitation by genetic engineering and, for instance, by transgenic overexpression of the respective human ligand, growth factor, cytokine or receptor within the murine organism33,35.
As a significant influence of chronic inflammation on the local secretion of chemokines, the endothelial expression of adhesion molecules and the subsequent accumulation of lymphocytes has been described in numerous publications38,39,40, the choice of a suitable experimental model of pulmonary inflammation represented a critical step while establishing the above described protocol and might be adapted dependent on the clinical context of each individual study. The here selected setting of papain-induced allergic lung inflammation represents a well-described experimental model, which is based on the capacity of the locally administered cysteine protease papain to irritate the airway epithelium and trigger the subsequent release of alarmins27,41,42. Interestingly, accidental exposition to papain is known to cause asthma development in humans as well43. Dependent on the inhaled dose of papain, exposed mice show an accumulation of innate and adaptive immune cells and elevated levels of type 2 cytokines in the lung27. While dose escalation turned out to result in an enlargement of airspaces (classic histological phenomenon of COPD) and a destruction of blood vessel walls with subsequent hemorrhage, moderate dosing schedules went along with a prominent pulmonary eosinophilia and thus mimicked an important histological feature of human asthma27. As our purpose was to use this experimental model to generate an inflammatory context for the analysis of pulmonary immune cell homing, we carefully tried to avoid papain-induced damage of blood vessels, which might otherwise result in an unphysiological extravasation of human immune cells due to disturbed endothelial integrity. Accordingly, we selected an intermediate dose of 50 µg of papain per mouse per day in the above described protocol. Although development of papain-induced airway inflammation also occurs in the absence of B and T lymphocytes, lung infiltrating adaptive immune cells are known to impact significantly on the course of disease27,42. For instance, regulatory T cells could be identified as potent regulators of papain-induced airway eosinophilia27. In perspective, the active involvement of pulmonary lymphocytes in the inflammatory pathogenesis of papain-exposed mice implicates that the above described protocol might in addition enable to analyze the functional capacity of adoptively transferred and successfully lung-accumulated human immune cells to modulate lung pathology (e.g., via flow cytometric acquisition of eosinophilic counts in BAL). Furthermore, an additionally performed intranasal administration of selected disease-relevant human chemokines just prior to intravascular cell transfer might allow to analyze the impact of these humoral mediators on the lung homing process of distinct human immune cell populations in an in vivo setting. Likewise, the effect of inhibitors on the lung homing process and, subsequently, on inflammatory lung pathology can be studied.
A particular advantage of the here introduced experimental procedure is the precise tracking and localization of lung infiltrating human immune cells by the high-end imaging quality of light-sheet fluorescence microscopy. Recent studies already demonstrated that the technique of light-sheet fluorescence microscopy represents a valuable experimental tool for analysis of intestinal immune cell homing in translational IBD research and is able to overcome the main limitations of conventional immunofluorescence microscopy and flow cytometry24,32. While analyses based on conventional microscopy are usually restricted to a very small and potentially not representative area of the organ and flow cytometry does not take into account the aspect of tissue organization at all, light-sheet fluorescence microscopy of chemically cleared tissue allows an increased penetration depth without major loss of resolution and thus enables a 3D reconstruction of rather large organ sections (up to 1.5 cm x 1.5 cm)24,28,44. For sure, the quality and validity of acquired 3D reconstructions critically depend on the performance of tissue clearance. In the here described setting, we included a slightly adopted version of the recently published protocol for ECi-based tissue clearing44. Compared to other well established strategies for solvent-based tissue clearing26,45,46, the ECi-based protocol combines the advantages of excellent clearing properties, low toxicity of used reagents and a moderate time requirement44. As the capacity of standard light-sheet fluorescence microscopy to resolve finest tissue structures like alveolar capillaries is still limited even under optimal clearing conditions28, it might somehow be difficult to reliably differentiate between intravascular and already extravasated immune cells. The inclusion of an additional standard fluorescence-based blood vessel staining into the here described protocol would most probably not be able to fully overcome this limitation. Thus, studies with a particular focus on the behavior of immune cells within the pulmonary microcirculation or their diapedesis might preferentially take into account a recently published and very elegant protocol for intravital lung imaging based on 2-photon microscopy47. In this experimental setting, a thoracic window was implanted into anesthetized and ventilated mice, through which the stabilized lung can be monitored by a resonant-scanning 2-photon microscopy module, allowing high-resolution imaging throughout the respiratory cycle upon preserved ventilation and perfusion47,48. This technique has been successfully applied in various studies and was able to visualize for instance intrapulmonary platelet biogenesis and the lung entrance of circulating tumor cells49,50. However, besides the requirement of sophisticated instrumental equipment (e.g., mouse ventilation system) and the need of extensive invasive manipulations in living animals (implantation of the thoracic window and intravital microscopy), the main limitation of this live lung microscopy system is the confined z-axis penetration. The performed lung surface imaging only allows analyzing the outer 100 µm of subpleural lung tissue47,48. Dependent on the scientific context, it might be a valuable option to implement 2-photon microscopy of the explanted lungs as an additional procedure into the above described protocol. Analyzing the same lung first by 2-photon microscopy and afterwards by light-sheet fluorescence microscopy might represent a strategy to obtain a quantitative overview of the distribution and localization of human immune cells within the murine lung (light-sheet fluorescence microscopy) and, at the same time, gain more detailed insights into cellular or microvascular processes (2-photon microscopy). Indeed, 2-photon microscopy of murine tracheal explants represents an established imaging tool for analyzing immune cell behavior in the context of experimentally induced lung inflammation51,52. Instead of visualizing small pulmonary capillaries, the successful extravasation and lung tissue infiltration of the transferred human T cells in our experimental model can also be proven by detecting fluorescently labeled human cells within the BAL of recipient animals via flow cytometry as exemplarily depicted in Figure 2E. In general, analyses of the cellular BAL compartment represent a well-established method to characterize the pulmonary immune cell influx in the context of inflammatory respiratory diseases31. Finally, another flow cytometric strategy for quantifying the extravasation of adoptively transferred immune cells within lung tissue was described by Galkina et al. (2005)53 and could potentially be combined with the here described protocol. In the study by Galkina et al., a single intravenous injection of a fluorescence-conjugated anti-CD8 antibody shortly before resection of the lung resulted in an exclusive and complete labeling of before transferred CD8+ T cells within the lung vascular compartment, while already extravasated CD8+ T cells within the lung interstitium remained unstained. Subsequent analyses of in vivo labeled pulmonary immune cells were performed flow cytometrically after ex vivo digestion of lung tissue53. Of course, the process of tissue digestion means that the anatomic separation between the intra- and extravascular lung compartment is abrogated and, thus, there is a general risk of false-positive labeling of interstitial immune cells due to antibody leakage between both compartments. This risk can only be minimized by performing the tissue digestion in the presence of saturating amounts of unlabeled antibody53 or, potentially, by replacing flow cytometric analysis by light-sheet fluorescence microscopy, which makes it possible to completely avoid the destruction of tissue structure.
In summary, the here introduced combination of in vivo homing of fluorescently labeled primary immune cells and subsequent light-sheet fluorescence microscopy is able to reliably identify, quantify and localize lung accumulated human immune cells in an experimental mouse model of pulmonary inflammation.
Divulgations
The authors have nothing to disclose.
Acknowledgements
The authors gratefully acknowledge funding by the DFG Collaborative Research Centers SFB 1181 and TRR 241. The Optical Imaging Centre Erlangen (OICE) and in particular Ralf Palmisano, Philipp Tripal and Tina Fraaß (Project Z2 of the DFG CRC 1181) are acknowledged for expert technical support for light-sheet fluorescence microscopic imaging.
Materials
| Agarose NEEO Ultra | Carl Roth GmbH + Co. KG, Karlsruhe, Germany | 2267.4 | |
| AlexaFlour594 anti-human CD45 antibody | BioLegend, San Diego, USA | 304060 | |
| Ammonium chloride | Carl Roth GmbH + Co. KG, Karlsruhe, Germany | K2981 | |
| Cannula 21 G | Becton, Dickinson and Company, Franklin Lakes, USA | 301300 | |
| Cell proliferation dye eflour670 | eBioscience Inc., San Diego, USA | 65-0840-85 | |
| CD4 MicroBeads, human | Miltenyi Biotech GmbH, Bergisch-Gladbach, Germany | 130-045-101 | |
| EDTA (ethylenediaminetetraacetic acid) | Carl Roth GmbH + Co. KG, Karlsruhe, Germany | 8043.1 | |
| Potassium-EDTA blood collection tube, 9 ml | Sarstedt AG & Co., Nümbrecht, Germany | 21066001 | |
| Ethly cinnamate (ECi) | Sigma-Aldrich, Steinheim, Germany | 112372-100G | |
| Ethanol ≥ 99.5 % (EtOH) | Carl Roth GmbH + Co. KG, Karlsruhe, Germany | 5054.3 | |
| FBS (fetal bovine serum) Good Forte | PAN-Biotech GmbH, Aidenbach, Germany | P40-47500 | |
| Filter 100 µm | VWR International Germany GmbH, Darmstadt, Germany | 732-2758 | |
| Imaris Image Analysis Software 9.0.2 | Bitplane AG, Zurich, Switzerland | n.a. | |
| ImspectorPro software | Abberior Instruments GmbH, Göttingen, Germany | n.a. | |
| Ketamin | Inresa Arzneimittel GmbH, Freiburg, Germany | 3617KET-V | |
| LaVision UltraMicroscope II | LaVision BioTec GmbH, Bielefeld, Germany | n.a. | |
| MACS MultiStand | Miltenyi Biotech GmbH, Bergisch-Gladbach, Germany | 130-042-303 | |
| Multifly cannula 20 G | Sarstedt AG & Co., Nümbrecht, Germany | 851638035 | |
| 30 G needle | B. Braun Melsungen AG, Melsungen, Hessen, Germany | 9161502 | |
| Neubauer counting chamber | neoLab Migge GmbH, Heidelberg, Germany | C-1003 | |
| Pattex Glue | Henkel AG & Co, Düsseldorf, Germany | PSK1C | |
| LS column | Miltenyi Biotech GmbH, Bergisch-Gladbach, Germany | 130-042-401 | |
| Lymphocyte Separation Media (Density 1,077 g/ml) | anprotec | AC-AF-0018 | |
| RPMI medium | (Gibco) Life Technologies GmbH, Darmstadt, Germany | 61870-010 | |
| Papain | Merck | 1,071,440,025 | |
| PBS Dulbecco (phosphate buffered saline) | Biochrom GmbH, Berlin, Germany | L182-10 | |
| PerCP/Cy5.5 anti-human CD4 | BioLegend, San Diego, USA | 317428 | |
| PerCP/Cy5.5 mouse IgG2b, κ isotype Ctrl | BioLegend, San Diego, USA | 400337 | |
| PFA (paraformaldehyde) | Carl Roth GmbH + Co. KG, Karlsruhe, Germany | 0335.1 | |
| Potassium hydrogen carbonate | Carl Roth GmbH + Co. KG, Karlsruhe, Germany | P7481 | |
| Serological pipette 10 ml | Sarstedt AG & Co., Nümbrecht, Germany | 86.1254.001 | |
| Syringe 1 ml | B. Braun Melsungen AG, Melsungen, Hessen, Germany | 9166017V | |
| Syringe 5 ml | Becton, Dickinson and Company, Franklin Lakes, USA | 260067 | |
| Syringe 20 ml | Becton, Dickinson and Company, Franklin Lakes, USA | 260069 | |
| Tube 1.5 ml | Sarstedt AG & Co., Nümbrecht, Germany | 72,706,400 | |
| Tube 2 ml | Sarstedt AG & Co., Nümbrecht, Germany | 72.695.400 | |
| Tube 2 ml, brown | Sarstedt AG & Co., Nümbrecht, Germany | 72,695,001 | |
| Tube 15 ml | Sarstedt AG & Co., Nümbrecht, Germany | 62.554.502 | |
| Tube 50 ml | Sarstedt AG & Co., Nümbrecht, Germany | 62.547.254 | |
| QuadroMACS Separator | Miltenyi Biotech GmbH, Bergisch-Gladbach, Germany | 130-090-976 | |
| Xylazin (Rompun 2%) | Bayer Vital GmbH, Leverkusen, Germany | KPOBD32 |
References
- Medoff, B. D., Thomas, S. Y., Luster, A. D. T cell trafficking in allergic asthma: the ins and outs. Annual Review of Immunology. 26, 205-232 (2008).
- Baraldo, S., Lokar Oliani, K., Turato, G., Zuin, R., Saetta, M. The Role of Lymphocytes in the Pathogenesis of Asthma and COPD. Current Medicinal Chemistry. 14 (21), 2250-2256 (2007).
- Mikhak, Z., Strassner, J. P., Luster, A. D. Lung dendritic cells imprint T cell lung homing and promote lung immunity through the chemokine receptor CCR4. Journal of Experimental Medicine. 210 (9), 1855-1869 (2013).
- Thomas, S. Y., Banerji, A., Medoff, B. D., Lilly, C. M., Luster, A. D. Multiple chemokine receptors, including CCR6 and CXCR3, regulate antigen-induced T cell homing to the human asthmatic airway. Journal of Immunology. 179 (3), 1901-1912 (2007).
- Katchar, K., Eklund, A., Grunewald, J. Expression of Th1 markers by lung accumulated T cells in pulmonary sarcoidosis. Journal of Internal Medicine. 254 (6), 564-571 (2003).
- Wu, Z., et al. Mast cell FcepsilonRI-induced early growth response 2 regulates CC chemokine ligand 1-dependent CD4+ T cell migration. Journal of Immunology. 190 (9), 4500-4507 (2013).
- Hart, P. H. Regulation of the inflammatory response in asthma by mast cell products. Immunology, Cell Biology. 79 (2), 149-153 (2001).
- Bice, J. B., Leechawengwongs, E., Montanaro, A. Biologic targeted therapy in allergic asthma. Annals of Allergy & Asthma & Immunology. 112 (2), 108-115 (2014).
- Barnes, N., et al. A randomized, double-blind, placebo-controlled study of the CRTH2 antagonist OC000459 in moderate persistent asthma. Clinical & Experimental Allergy. 42 (1), 38-48 (2012).
- Medoff, B. D., et al. CD11b+ myeloid cells are the key mediators of Th2 cell homing into the airway in allergic inflammation. Journal of Immunology. 182 (1), 623-635 (2009).
- Oeser, K., Maxeiner, J., Symowski, C., Stassen, M., Voehringer, D. T. T cells are the critical source of IL-4/IL-13 in a mouse model of allergic asthma. Allergy. 70 (11), 1440-1449 (2015).
- Freeman, C. M., Curtis, J. L., Chensue, S. W. CC chemokine receptor 5 and CXC chemokine receptor 6 expression by lung CD8+ cells correlates with chronic obstructive pulmonary disease severity. The American Journal of Pathology. 171 (3), 767-776 (2007).
- Kallinich, T., et al. Chemokine-receptor expression on T cells in lung compartments of challenged asthmatic patients. Clinical & Experimental Allergy. 35 (1), 26-33 (2005).
- Vasakova, M., et al. Bronchoalveolar lavage fluid cellular characteristics, functional parameters and cytokine and chemokine levels in interstitial lung diseases. Scandinavian Journal of Immunology. 69 (3), 268-274 (2009).
- Campbell, J. J., et al. Expression of chemokine receptors by lung T cells from normal and asthmatic subjects. Journal of Immunology. 166 (4), 2842-2848 (2001).
- Halwani, R., et al. IL-17 Enhances Chemotaxis of Primary Human B Cells during Asthma. PLoS One. 9 (12), 114604 (2014).
- Agostini, C., et al. Cxcr3 and its ligand CXCL10 are expressed by inflammatory cells infiltrating lung allografts and mediate chemotaxis of T cells at sites of rejection. The American Journal of Pathology. 158 (5), 1703-1711 (2001).
- Ainslie, M. P., McNulty, C. A., Huynh, T., Symon, F. A., Wardlaw, A. J. Characterisation of adhesion receptors mediating lymphocyte adhesion to bronchial endothelium provides evidence for a distinct lung homing pathway. Thorax. 57 (12), 1054-1059 (2002).
- Radeke, H. H., Ludwig, R. J., Boehncke, W. H. Experimental approaches to lymphocyte migration in dermatology in vitro and in vivo. Experimental Dermatology. 14 (9), 641-666 (2005).
- Miles, A., Liaskou, E., Eksteen, B., Lalor, P. F., Adams, D. H. CCL25 and CCL28 promote alpha4 beta7-integrin-dependent adhesion of lymphocytes to MAdCAM-1 under shear flow. American Journal of Physiology-Gastrointestinal and Liver Physiology. 294 (5), 1257-1267 (2008).
- Zundler, S., et al. The alpha4beta1 Homing Pathway Is Essential for Ileal Homing of Crohn’s Disease Effector T Cells In vivo. Inflammatory Bowel Diseases. 23 (3), 379-391 (2017).
- Verbist, K. C., Cole, C. J., Field, M. B., Klonowski, K. D. A role for IL-15 in the migration of effector CD8 T cells to the lung airways following influenza infection. Journal of Immunology. 186 (1), 174-182 (2011).
- Kopf, M., Abel, B., Gallimore, A., Carroll, M., Bachmann, M. F. Complement component C3 promotes T-cell priming and lung migration to control acute influenza virus infection. Nature Medicine. 8 (4), 373-378 (2002).
- Zundler, S., et al. Three-Dimensional Cross-Sectional Light-Sheet Microscopy Imaging of the Inflamed Mouse Gut. Gastroenterology. 153 (4), 898-900 (2017).
- Mzinza, D. T., et al. Application of light sheet microscopy for qualitative and quantitative analysis of bronchus-associated lymphoid tissue in mice. Cellular & Molecular Immunology. , (2018).
- Erturk, A., Lafkas, D., Chalouni, C. Imaging cleared intact biological systems at a cellular level by 3DISCO. Journal of Visualized Experiments. (89), 51382 (2014).
- Morita, H., et al. An Interleukin-33-Mast Cell-Interleukin-2 Axis Suppresses Papain-Induced Allergic Inflammation by Promoting Regulatory T Cell Numbers. Immunity. 43 (1), 175-186 (2015).
- Mann, L., Klingberg, A., Gunzer, M., Hasenberg, M. Quantitative Visualization of Leukocyte Infiltrate in a Murine Model of Fulminant Myocarditis by Light Sheet Microscopy. Journal of Visualized Experiments. (123), 55450 (2017).
- Mercer, R. R., et al. Extrapulmonary transport of MWCNT following inhalation exposure. Particle and Fibre Toxicology. 10, 38 (2013).
- Minton, C., et al. Demonstration of microvessel networks and endothelial cell phenotypes in the normal murine lung. Journal of Nippon Medical School. 72 (6), 314-315 (2005).
- Van Hoecke, L., Job, E. R., Saelens, X., Roose, K. Bronchoalveolar Lavage of Murine Lungs to Analyze Inflammatory Cell Infiltration. Journal of Visualized Experiments. (123), 55398 (2017).
- Fischer, A., et al. Differential effects of alpha4beta7 and GPR15 on homing of effector and regulatory T cells from patients with UC to the inflamed gut in vivo. Gut. 65 (10), 1642-1664 (2016).
- Shultz, L. D., Ishikawa, F., Greiner, D. L. Humanized mice in translational biomedical research. Nature Reviews Immunology. 7 (2), 118-130 (2007).
- Brehm, M. A., Jouvet, N., Greiner, D. L., Shultz, L. D. Humanized mice for the study of infectious diseases. Current Opinion in Immunology. 25 (4), 428-435 (2013).
- Wege, A. K. Humanized Mouse Models for the Preclinical Assessment of Cancer Immunotherapy. BioDrugs. , (2018).
- Jespersen, H., et al. Clinical responses to adoptive T-cell transfer can be modeled in an autologous immune-humanized mouse model. Nature Communications. 8 (1), 707 (2017).
- Schloder, J., Berges, C., Luessi, F., Jonuleit, H. Dimethyl Fumarate Therapy Significantly Improves the Responsiveness of T Cells in Multiple Sclerosis Patients for Immunoregulation by Regulatory T Cells. International Journal of Molecular Sciences. 18 (2), (2017).
- Murdoch, C., Finn, A. Chemokine receptors and their role in inflammation and infectious diseases. Blood. 95 (10), 3032-3043 (2000).
- Rivera-Nieves, J., Gorfu, G., Ley, K. Leukocyte adhesion molecules in animal models of inflammatory bowel disease. Inflammatory Bowel Diseases. 14 (12), 1715-1735 (2008).
- Zundler, S., Neurath, M. F. Novel Insights into the Mechanisms of Gut Homing and Antiadhesion Therapies in Inflammatory Bowel Diseases. Inflammatory Bowel Diseases. 23 (4), 617-627 (2017).
- Halim, T. Y., Krauss, R. H., Sun, A. C., Takei, F. Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity. 36 (3), 451-463 (2012).
- Kamijo, S., et al. IL-33-mediated innate response and adaptive immune cells contribute to maximum responses of protease allergen-induced allergic airway inflammation. Journal of Immunology. 190 (9), 4489-4499 (2013).
- Milne, J., Brand, S. Occupational asthma after inhalation of dust of the proteolytic enzyme, papain. British Journal of Industrial Medicine. 32 (4), 302-307 (1975).
- Klingberg, A., et al. Fully Automated Evaluation of Total Glomerular Number and Capillary Tuft Size in Nephritic Kidneys Using Lightsheet Microscopy. Journal of the American Society of Nephrology. 28 (2), 452-459 (2017).
- Ariel, P. A beginner’s guide to tissue clearing. The International Journal of Biochemistry, Cell Biology. 84, 35-39 (2017).
- Richardson, D. S., Lichtman, J. W. Clarifying Tissue Clearing. Cell. 162 (2), 246-257 (2015).
- Looney, M. R., et al. Stabilized imaging of immune surveillance in the mouse lung. Nature Methods. 8 (1), 91-96 (2011).
- Looney, M. R., Bhattacharya, J. Live imaging of the lung. Annual Review of Physiology. 76, 431-445 (2014).
- Lefrancais, E., et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature. 544 (7648), 105-109 (2017).
- Headley, M. B., et al. Visualization of immediate immune responses to pioneer metastatic cells in the lung. Nature. 531 (7595), 513-517 (2016).
- Hammad, H., et al. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nature Medicine. 15 (4), 410-416 (2009).
- Bose, O., et al. Mast cells present protrusions into blood vessels upon tracheal allergen challenge in mice. PLoS One. 10 (3), 0118513 (2015).
- Galkina, E., et al. Preferential migration of effector CD8+ T cells into the interstitium of the normal lung. Journal of Clinical Investigation. 115 (12), 3473-3483 (2005).
