Since the cannula is placed just dorsal to CA1, the dorsal aspect of the CA1 is more proximal to the microscope if compared to the ventral one. The alveus is the most proximal structure followed in order by stratum oriens (SO), stratum pyramidale (SP), stratum radiatum (SR) and stratum lacunosum moleculare (SLM), the most distal layer (Figures 1B, C). Longitudinal 2P imaging of neuronal structure with subcellular resolution and of activity-evoked plasticity with cellular resolution are presented as representative results. To excite the fluorophores green fluorescent protein (GFP), d2Venus and TurboFP635, a femtosecond pulsed Ti:Sapphire laser tuned to 920 nm is used and the average laser power is adjusted to 5-25 mW at the sample. The different emission wavelengths are separated using emission filters and different photomultiplier tubes.
Longitudinal imaging of dendritic structure and dendritic spines dynamics.
To sparsely label hippocampal PNs and visualize their structure, a Thy1-GFP transgenic mouse line (M line) is used. Thy1-GFP mice express cytoplasmic enhanced GFP under the control of the Thy1 promoter in a sparse, random population of PNs25. Generally, the major axis of CA1 PNs is roughly perpendicular to the XY-imaging plane (Figures 1B, Figure 2A and Supplementary Movie 1). Basal dendrites extend in the SO, from the soma towards the cannula, whereas apical and apical tuft dendrites extend distally to the cannula, in the opposite direction (Supplementary Movie 1). Since the preparation leaves the deepest fibers of the alveus intact, a few fluorescent fibers traversing the field of view, just beneath the cannula, should be visible (Figure 2A and Movie 1). The preparation allows imaging of PN dendrites with spine resolution (Figure 2 A-C). To image dendrites and dendritic spines, a 25X 1.0 NA, 4 mm WD, water immersion commercial objective lens is used.
For longitudinal tracking, several brain regions within the field of view of the cannula are defined during the first imaging session. Each region corresponds to an area of approximately 240 x 240 µm and contains between 1 and 7 dendritic segments (Figure 2B). These regions are manually mapped to a low magnification three-dimensional stack showing the pattern of GFP expression in the volume below the imaging cannula (Figure 2A). Then, 1 µm z-step image stacks of CA1 PN basal dendrites are acquired at different time intervals (from 24 h to 3 days) for up to about 14 days (Figure 2C). Longer imaging durations and intervals are possible26. Each imaging session lasts approximately 60 to 90 min. Although most images are of dendritic spines in the SO, it is also possible to image dendritic spines in the oblique dendrites of SR (Figures 2D-F). In addition to spine density, this method enables the study of spine dynamics by quantifying their survival, gain and loss rates26,27,28,29,30,31,32. To score and track dendritic spines over time (Figure 2C), a custom MATLAB interface is used. This enables the alignment of the image stacks acquired at different time points and supports manual labeling of dendrites and spines while tracking dendritic lengths and spine positions over time26. Importantly, this method can be used to distinguish (per each time point, excluding the first one) between pre-existing and newborn dendritic spines. This is important as the different classes of dendritic spines are thought to have different roles in memory acquisition and retention33.
Longitudinal imaging of activity-evoked plasticity.
To image activity-evoked plasticity in CA1 PNs, the dorsal CA1 hippocampal area is injected with a viral vector expressing green fluorescent destabilized d2Venus via an enhanced form of the synaptic activity-responsive element (E-SARE) within the Arc enhancer/promoter and red fluorescent TurboFP635 via the ePGK promoter34. This allows for imaging levels of activity-evoked plasticity of hundreds of CA1 PNs in each animal35. Given the very dense labeling of PNs, it is generally not possible to resolve the dendrites of CA1 PNs (Supplementary Movie 2).
To image the somata of CA1 PNs, a 40X 0.8 NA, 3 mm WD, water immersion objective lens is used. For longitudinal tracking, 1 to 9 brain regions are defined per mouse during the first imaging session. Each region corresponds to an area of approximately 300 x 300 µm and contains between 50 and 150 cells (Figure 3A). These regions are manually mapped to local tissue landmarks visible at a lower magnification. Then, 3 µm z-step image stacks are acquired, which encompass the SP of CA1 PNs (Figure 3A and Supplementary Movie 2) at different time intervals (from 24 h to 6 days) for up to about 30 days. Each imaging session lasts approximately 60 to 90 min. E-SARE activation peaks 6 to 8 h after an exposure to a new or enriched environment (EE, Figure 3B) and decays over the course of a few days. Thus, we generally image 6 to 8 h after experience and allow for 5 days between imaging sessions35.
To quantify d2Venus and TurboFP635 fluorescence values, a circular region-of-interest 4.64 µm in diameter is drawn, which is smaller than a neural cell body, centered to the cell soma. We then progress to the next time point, score the soma of the same cell in the same way, and iterate this procedure for all time points and all visible cells in the longitudinal dataset. The mean value of each neuron’s (activity-dependent) d2Venus emission is normalized by its mean (activity-independent) TurboFP635 emission. This method enables the investigation of long-term dynamics of ensemble plasticity of CA1 PNs35 (Figure 3C).
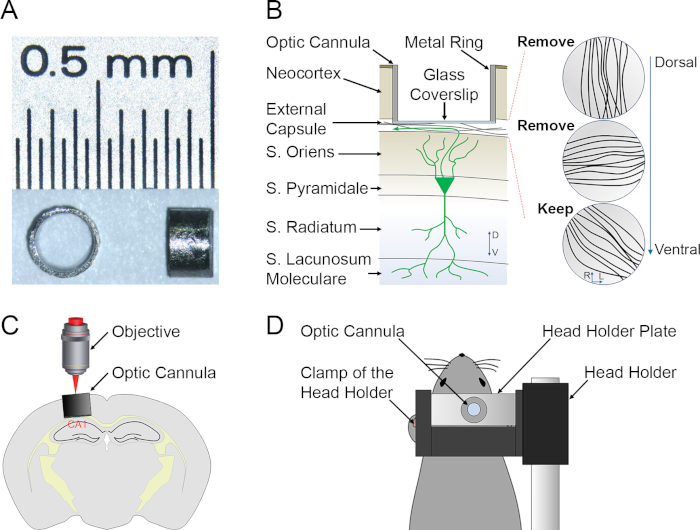
Figure 1: Preparation for in vivo deep brain optical imaging. (A). Top (left) and side (right) views of example imaging cannuals. Imaging cannulas have a transparent glass bottom to allow optical access to the hippocampus. (B). Schematic description of the preparation highlighting the relative position of the imaging cannula, a CA1 PN and the three layers of fibers from dorsal to ventral. (C, D). Schematic description of the imaging setup (C) and of the animal fixation (D) during an imaging session. Please click here to view a larger version of this figure.
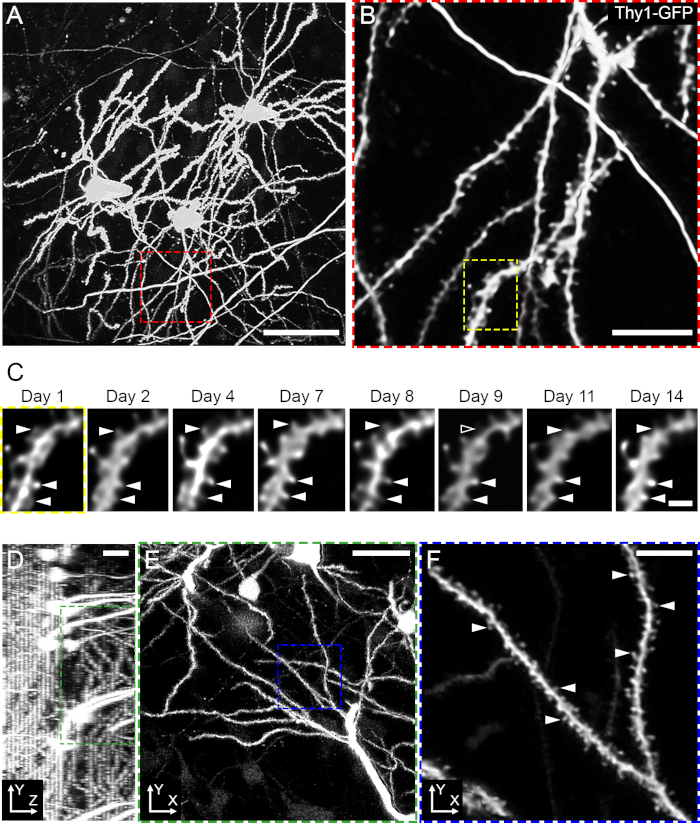
Figure 2: Longitudinal imaging of dendritic structure and dendritic spines dynamics. (A). 2P image stack (Maximum Intensity Projection (MIP) of 59 image planes, 2 µm z-spacing) of neurons and dendrites labelled by GFP in a live Thy1-GFP mouse. (B). Higher magnification (MIP of 53 image planes, 1 µm z-spacing) detailing basal dendrites located in SO. (C). Time-lapse image sequence of a dendritic segment imaged over 14 days. Arrowheads indicate dendritic spines tracked over 14 days. (D, E). 2P image stack of neurons and dendrites labelled by GFP in a live Thy1-GFP mouse; (D) ZY projection (31 image planes, 3 µm z-spacing) and (E) XY projections (17 image planes, 3 µm z-spacing). (F). Higher magnification (single image plane) detailing apical dendrites and dendritic spines located in SR. Arrowheads indicate dendritic spines. Excitation: 920 nm; emission peak: 510 nm. Scale bars: A, 50 µm; B, 10 µm; C, 2 µm; D and E, 15 µm; F, 4 µm. Please click here to view a larger version of this figure.
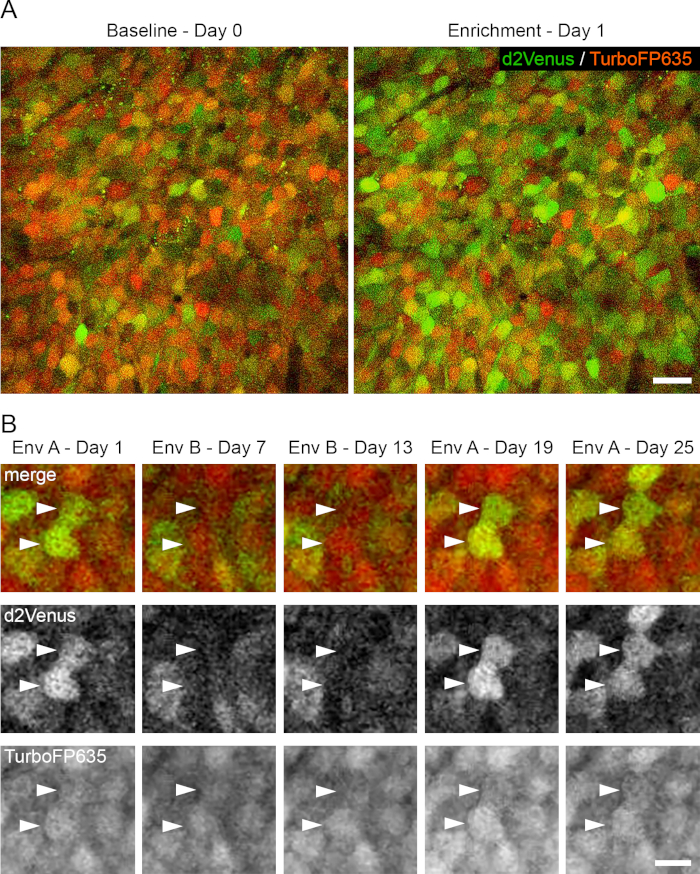
Figure 3: Longitudinal imaging of activity-evoked plasticity. (A). 2P images (single image planes) from a live mouse, showing the same cells on Baseline Day 0 and after EE on Day 1. (B). 2P image stacks (MIPs of 4-6 image planes, 3 µm z-spacing) showing E-SARE activation patterns specific for environment A (Days 1, 19 and 25) and environment B (Days 7 and 13). Green: d2Venus fluorescence. Red: TurboFP635 fluorescence. Excitation: 920 nm; d2Venus emission peak: 530 nm; TurboFP635 emission peak: 635nm. Scale bars: A, 20 µm; B, 10 µm. This figure has been modified from Attardo et al., 201835. Please click here to view a larger version of this figure.
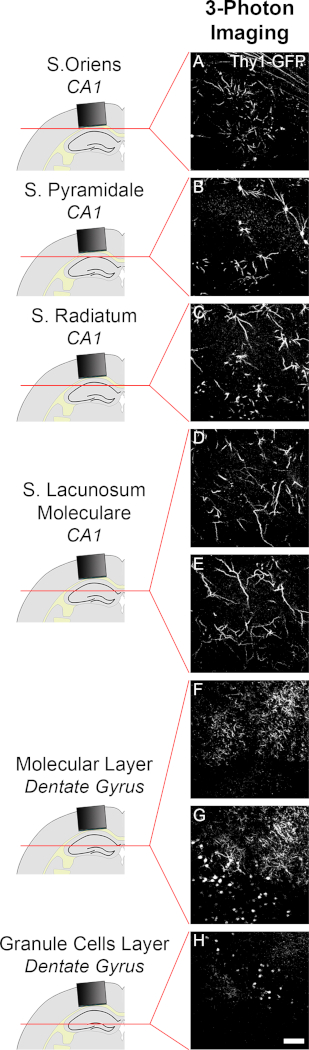
Figure 4: Imaging of neuronal structure in hippocampal DG using three-photon (3P) microscopy. (A-H). 3P images (single image planes) of neurons and dendrites labelled by GFP in a live Thy1-GFP mouse detailing (A-E) PNs in the CA1 and (F-H) granule cells in the DG. Excitation: 1400 nm; emission peak: 510 nm. Scale bar: 40 µm. Please click here to view a larger version of this figure.
Supplementary Movie 1: Imaging field of view in a live Thy1-GFP mouse. 2P image stack (83 image planes, 7 µm z-spacing) of neurons and dendrites labelled by GFP (white) in a live Thy1-GFP mouse extending from the bottom of the cannula to SLM. To account for the decay of fluorescence signal with increasing depth, we used a non-linear gradient of photomultiplier tubes’ gain. Please click here to download this file.
Supplementary Movie 2: Imaging of activity-evoked plasticity. 2P image stack (28 image planes, 3 µm z-spacing) of neurons expressing E-SARE reporter of IEG expression in a live mouse encompassing SP. Green: d2Venus fluorescence. Red: TurboFP635 fluorescence. Please click here to download this file.
