지난 2년 동안 응집된 유기 분자로 만들어진 고형물및 막은 광전자 장치에서 여러 응용 분야에서 발견되었습니다. 이러한 재료를 기반으로 하는 장치는 작은 무게, 유연성, 낮은 전력 소비 및 잉크젯 인쇄를 사용하여 저렴한 생산을 위한 잠재력을 포함하여 많은 매력적인 특성을 가지고 있습니다. 유기 발광 다이오드(OLED)를 기반으로 하는 디스플레이는 휴대 전화, 노트북, 텔레비전 세트 및 기타 전자 기기1,2,2,3,,4에대한 최첨단으로 액정 디스플레이를 대체하고 있습니다. 조명 응용 프로그램에 대한 OLED의 중요성은 향후4년 동안 증가 할 것으로 예상된다. 유기 태양광 장치의 성능은 꾸준히 개선되고 있으며, 최근 단일 접합 유기 태양전지에 대해 16% 이상의 전력 변환 효율이보고되고 있다 5. 유기 재료는 또한 광섬유 통신과 같은 다른 기술을 방해 할 수있는 잠재력을 가지고 있으며, 그 사용은 15 THz 및6,7이상의 매우 높은 대역폭을 가진 전기 광학 변조기의 개발을 가능하게합니다.
광전자공학 의 응용 제품을 위한 고체 분자 물질을 최적화하는 데 있어 주요 과제는 일반적으로 그 특성이 재료의 나노 스케일 구조에 크게 의존한다는 것입니다. 생산 공정은 화학 증착, 광학 활성 분자의8 템플릿을 다른 물질 (즉, 폴리머 매트릭스9,,10),열 어닐링11,,12등과 같은 제어 된 성장 기술을 사용하여 재료의 나노 구조를 어느 정도 정의 할 수 있습니다. 그러나, 나노 스케일 장애는 대부분의 분자 물질에 내재되어 있으며 일반적으로 완전히 제거 될 수 없습니다. 따라서 장애가 재료의 특성에 미치는 영향을 이해하고 최적의 성능을 위해 설계하는 방법을 찾는 것은 유기 광전자 재료의 합리적인 설계에 필수적입니다.
분자 물질에 있는 무질서의 정도는 일반적으로 밴드 이론에 의해 기술될 수 있는 전자 구조물을 가진 주기적인 결정 구조물의 섭동으로 취급하기 위하여 너무 중대합니다. 한편, 벌크 물질 또는 필름의 특성을 재현하기 위해 시뮬레이션에 포함되어야 하는 분자의 수는 밀도 기능이론(DFT)13,,14 및 시간 의존밀도 기능이론(TD-DFT)15,,16과같은 제1 원리 양자 화학적 방법을 사용하기에는 너무 크다. 광전자공학에 응용되는 유기 분자는 전형적으로 상대적으로 큰 π-컨쥬게이션 시스템을 갖는다; 많은 사람들이 또한 기증자 와 수락자 그룹이 있습니다. 이러한 분자에서 올바른 전하 전달 거동을 포착하는 것은 광전자적 특성을 계산하는 데 필수적이지만 TD-DFT17,,18,19,,20에서장거리 보정 하이브리드 기능을 사용하여만 수행할 수 있습니다., 이러한 기능을 사용하는 계산은 시스템의 크기에 따라 매우 선형적으로 확장되며, 현재는 ~10 4 원자 기준 함수를 사용하여 설명할 수 있는 개별 유기 분자 또는 작은 분자 응집체의 광전자적 특성을 모델링하는 데만 실용적입니다. 많은 수의 크로모포로 구성된 무질서한 재료를 설명할 수 있는 시뮬레이션 방법은 이러한 시스템을 모델링하는 데 매우 유용합니다.
분자 물자에 있는 분자 간 상호 작용의 크기는 수시로 물질을 구성하는 개별 분자 사이 에너지 매개변수 (eigenstate 에너지 또는 여기 에너지와 같은)에 있는 변이의 순서에 비교하거나 더 작습니다. 이러한 경우, 다중 스케일 모델링은 대형 장애 분자 시스템21,,22,,23의광전자 적 특성을 이해하고 최적화하는 가장 유망한 접근 방식이다. 이 접근법은 제1 원리 양자 화학 방법(일반적으로 DFT 및 TD-DFT)을 사용하여 물질을 구성하는 개별 분자의 특성을 정확하게 계산합니다. 벌크 분자 물질을 나타낼 수 있을 만큼 충분히 큰 재료 샘플의 Hamiltonian은 (아마도 주기적인 경계 조건을 사용하여) 개별 분자에 대해 계산된 매개 변수를 사용하여 구성됩니다. 이 해부니아니어는 큰 분자 골재, 박막 또는 벌크 분자 물질의 광전자 파라미터를 계산하는데 사용될 수 있다.
엑시톤 모델은 분자 물질의 흥분 상태가 엑시톤의기초로 표현되는 멀티 스케일 모델의 하위 클래스입니다 : 쿨롱 매력24,,25에의해 구속되는 전자 구멍 쌍. 많은 흥분 상태 프로세스를 모델링하는 경우, 전자와 구멍이 동일한 분자에 국한되어 있는 Frenkel excitons26만포함하기에 충분합니다. 전하 전달 엑시톤은, 여기서 전자 및 구멍이 상이한 분자 상이한 분자상에 국한되어, 일부 경우에 포함되어야 할 수도 있다(예를 들어, 공체 수용체 시스템에서 전하 분리를 모델링하는 경우)27,,28. 엑시톤 모델은 개별 분자에 대한 첫 번째 원리 계산만사용하여 파라메트화할 수 있는 다중 스케일 모델이지만, 여전히 분자간 상호 작용을 고려합니다. 그들이 설명 할 수있는 두 가지 기본 상호 작용 유형은 (a) 주위 분자에 전하 분포에 의해 분자에 전자 밀도의 정전기 편광을 가로 질러 지역화또는 전달하는 엑시톤의 능력을 특성화하는 분자 사이의 엑시토닉 커플링입니다. 우리는 이전에 이 두 요인이 광학 흡수 스펙트럼29 및 제1 극극성30과같은 분자 응집체의 광학 및 전기 광학 특성을 모델링하는 데 중요하다는 것을 보여주었습니다.
이 백서에서는 대형 분자 응집체 및 벌크 분자 물질의 광학 스펙트럼 및 기타 광전자적 특성을 계산하는 데 사용할 수 있는 엑시톤 모델을 파라메트화하기 위한 프로토콜을 제시합니다. 흥분제는 단단한 결합 해밀턴 으로 가정24,,25,
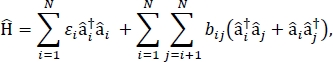
여기서θ i는 물질 내의 ith 분자의 여기 에너지인데, bij는 ith 분자와 jthth 분자 사이의 엑시토닉 커플링이며, âi† 및 âi는 각각 물질의 ith 분자상에서 의 한 분자에 대한 흥분 상태에 대한 생성 및 소멸 연산자이다. 흥분 성 해밀턴 매개 변수는 물질을 구성하는 개별 분자에서 수행되는 TD-DFT 계산을 사용하여 발견됩니다. 이러한 TD-DFT 계산에서, 재료의 다른 모든 분자에 대한 전하 분포는 분자의 전자 밀도의 정전기 편광을 고려하여 원자점 전하의 정전기 포함으로 표현됩니다. 개별 분자에 대한 여기 에너지, θi는TD-DFT 계산 출력에서 직접 가져온다. 상기 엑시토닉 커플링, bij,분자 들 간의 전이 밀도 큐브방법(31)을사용하여 계산되고, 가우시안(32)에서32 TD-DFT 계산의 출력으로부터 가져온 상호작용 분자에 대한 접지-흥분 상태 전이 밀도와 함께 다원다능 파함수 분석기(33)를 사용하여 후처리한다. Multiwfn 33 벌크 분자 고형물의 특성을 시뮬레이션하기 위해 주기적인 경계 조건을 Hamiltonian에 적용할 수 있습니다.
현재 프로토콜은 사용자가 Gaussian32 및 Multiwfn33 프로그램에 액세스할 수 있어야 합니다. 프로토콜은 Gaussian 16, 개정 B1 및 Multiwfn 버전 3.3.8을 사용하여 테스트되었지만 이러한 프로그램의 다른 최신 버전에서도 작동해야 합니다. 또한 이 프로토콜은 사용자 지정 C++ 유틸리티와 https://github.com/kocherzhenko/ExcitonicHamiltonian GNU 일반 공용 라이선스(버전 3)에 따라 제공되는 소스 코드인 사용자 지정 파이썬 2.7 및 Bash 스크립트를 사용합니다. 계산은 유닉스/Linux 제품군에서 운영 체제를 실행하는 컴퓨터에서 수행됩니다.
