Recent technological advances in genomics (transcriptomics) allow for comprehensive and precise analysis of the genome (transcriptome) in single cells1,2,3. However, single-cell proteomics technologies are lagging far behind but are just as important as genomics (transcriptomics) technologies4,5,6,7,8. Furthermore, protein abundance cannot necessarily be inferred from mRNA abundance9, and the proteome is more complex and dynamic than the transcriptome10. Given these challenges, a large number of mixed populations of cells (i.e., bulk cells) are generally used to generate comprehensive proteome data11,12,13. However, such bulk measurements average out stochastic variations of individual cells, thus obscure important cell-to-cell variability (i.e., cell heterogeneity)4,14. Limitations of such bulk measurements become even more severe when the cells of interest only account for a small portion of the total populations of cells (e.g., cancer stem cells within tumors at an early-stage cancer). Therefore, there is a huge knowledge gap between single-cell proteomics and genomics (transcriptomics).
Antibody-based immunoassays (e.g., flow or mass cytometry) are predominantly used for targeted proteomic analysis of single cells6,7,15,16,17,18. However, they suffer from low multiplex, limited specificity, and unavailability of antibodies for new proteins of interest. Mass spectrometry (MS)-based targeted proteomics has emerged as an alternative for accurate protein quantification because of its being antibody-free, high multiplex (≥150 proteins in a single analysis19), high quantitation accuracy (absolute amounts or concentrations), and high specificity and reproducibility (≤10% CV)20,21,22,23. Recent significant progress in sample preparation has made MS-based single-cell proteomics possible for quantitative analysis of highly abundant proteins from single human cells. However, MS-based single-cell proteomics is still at the early infancy stage. For example, the most advanced MS platform coupled with ultralow-flow RPLC flow rates can only allow label-free MS detection and quantification of ~670-870 proteins out of the total ≥12,000 proteins in single HeLa cells24,25.
Currently, there are six MS-based single-cell proteomics approaches available for analysis of single mammalian cells, in which four are for global proteomics (nanoPOTS: nanowell-based Preparation in One pot for Trace Samples26; iPAD-1: integrated proteome analysis device for single-cell analysis27; OAD (oil-air-droplet) chip-based single cell proteomic analysis28; SCoPE-MS: single cell proteomics by mass spectrometry29) and the other two are for targeted proteomics (cLC-SRM: carrier-assisted liquid chromatography (LC) – selected reaction monitoring (SRM)30; cPRISM-SRM: carrier-assisted high pressure, high-resolution separations with intelligent selection and multiplexing coupled to SRM31). However, all these approaches have technical drawbacks. nanoPOTS, iPAD-1, and OAD downscale sample processing volume to 2-200 nL and are not ready for broad benchtop applications26,27,28. For SCoPE-MS, a TMT (tandem mass tag) carrier is added after single-cell processing, so it cannot effectively prevent surface adsorption losses during sample processing when a single tube is used for single-cell processing29, resulting in low reproducibility with a correlation coefficient of only ~0.2-0.4 between replicates32. For cLC-SRM and cPRISM-SRM, using exogenous proteins as a carrier is more suitable for targeted proteomics because peptides from excessive exogenous proteins are frequently sequenced by MS/MS, which greatly reduces the chance for sequencing low abundant endogenous peptides30,31. Unlike global proteomics for relative quantification, the two targeted proteomics approaches can provide accurate or absolute protein analysis of small numbers of cells with high reproducibility using heavy isotope-labeled internal standards at known concentrations. When compared to cPRISM-SRM that requires prior high-resolution PRISM fractionation, resulting in many fraction samples that need to be analyzed, cLC-SRM has a significant advantage in sample throughput without fractionation and can simultaneously quantify hundreds of proteins in a single analysis but with relatively lower detection sensitivity30. Therefore, cLC-SRM is more accessible and should have broader utilities for accurate multiplexed protein analysis of small numbers of cells as well as mass-limited samples.
Herein we describe a detailed protocol to perform cLC-SRM for convenient targeted proteomics analysis of small numbers of human cells, including single cells. The protocol consists of the following major steps: cell sorting by FACS (fluorescence activated cell sorting), cell lysis and digestion processed in low-volume single polymerase chain reaction (PCR) tubes, LC-SRM data collection, and SRM data analysis using publicly available Skyline software (Figure 1). Its broad utility was demonstrated along with our previously well-established SRM assays by absolute targeted quantification of EGFR/MAPK pathway proteins in 1-100 MCF7 or MCF10A cells and determination of pathway protein copies per cell at a wide dynamic range of concentrations30. We anticipate that with the detailed protocol most proteomics researchers can readily implement cLC-SRM in their laboratories for accurate protein analysis of ultrasmall samples (e.g., rare tumor cells) to meet their project needs.
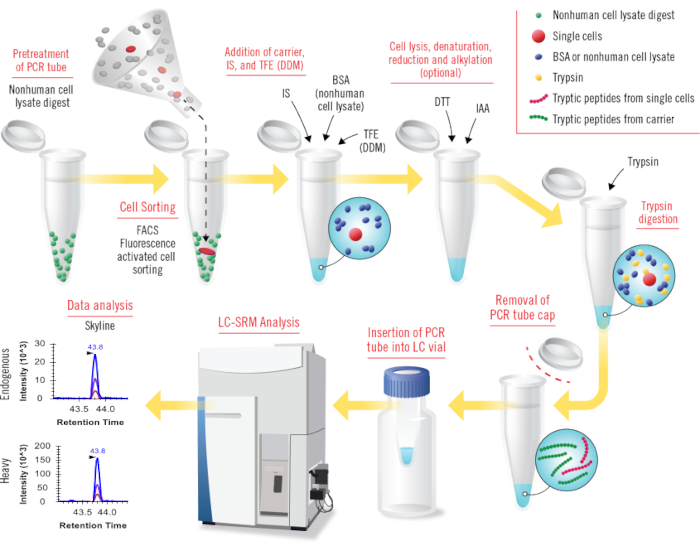
Figure 1: Overview of all steps in cLC-SRM (carried-assisted one-pot sample preparation coupled with liquid chromatography –selected reaction monitoring). Nonhuman cell lysate digests (e.g., Shewanella oneidensis) are used to pretreat PCR tubes for coating tube surface. Small numbers of human cells or single cells sorted by FACS are collected into pretreated PCR tubes. BSA protein (or nonhuman cell lysate proteome) carrier, heavy internal standard (IS), and TFE (or DDM) are added into sample tubes sequentially for facilitating cell lysis and reducing surface adsorption losses. Conceptually, the combined DDM and nonhuman cell lysate proteome carrier will work well for cLC-SRM. Cell lysis is conducted by sonication, and protein denaturation is achieved by heating at high temperature. DTT and IAA reagents are used for reduction and alkylation, respectively (this step is optional). Trypsin is added for digestion with much higher ratios of trypsin over protein amount than that for standard trypsin digestion. The cap of sample tube is removed and then PCR tube is inserted into LC vial for direct LC-SRM analysis. Collected SRM data are analyzed by using publicly available Skyline software. Please click here to view a larger version of this figure.
