NOTE: For the following protocol, transient transfection of protoplasts was performed, as described previously12. A brief description is given below.
1. Transient transfection of protoplasts
- Cut ~4 g of healthy leaves of Arabidopsis thaliana ecotype Columbia into 1 mm slices and transfer them to 20 mL of enzyme solution (1.5% cellulase; 0.4% macerozyme; 0.1% bovine serum albumin Fraction V; 0.4 M mannitol; 20 mM KCl; 20 mM 2-(N-morpholino)ethanesulfonic acid (MES), pH 5.7; 10 mM CaCl2).
- Vacuum-infiltrate the leaf slices followed by an incubation with agitation for 2 h at room temperature. Harvest the cells by centrifugation for 3 min at 100 × g.
- Wash the protoplasts with W5-solution (154 mM NaCl; 125 mM CaCl2; 5 mM KCl; 2 mM MES, pH 5.7) and resuspend them in MMG solution (0.4 M mannitol; 15 mM MgCl2; 4 mM MES, pH 5.7).
- Perform the transfection in an 8-well slide by osmotic shock in the presence of polyethyleneglycol (PEG) 4000. Mix 20 µL of the protoplast suspension with 5 µL of plasmid DNA (5 µg/µL) and 25 µL of PEG solution (0.2 M mannitol, 0.1 M CaCl2, 40% PEG 4000).
- Reverse the osmotic shock by gentle readjustment of the osmotic conditions.
NOTE: Besides the sample of interest, the expression of donor alone and acceptor alone is required to determine the spectral bleed-through of the donor and the acceptor, respectively. A fusion protein of the donor and the acceptor must be expressed too for calibration purposes. The fluorescent protein expression was under the control of a cauliflower mosaic virus 35S promoter (pCaMV35S). For all measurements, two confocal laser scanning microscopes (LSM1 and LSM2) were used. LSM1 has two types of detectors: for FRET measurements, the donor signal was detected by a GaAsP-detector, while FRET and acceptor emission were recorded with a photomultiplier. LSM2 has two photomultipliers, which were used for the detection of donor, FRET, and acceptor emission.
2. Laser-adjustment
NOTE: Here, 458 nm and 514 nm lines of an argon-ion laser have been applied for FRET analysis between enhanced cyan fluorescent protein (ECFP)- and enhanced yellow fluorescent protein (EYFP)-labeled proteins. For reproducible data acquisition, both lines were adjusted to similar intensity. This was achieved by either a transmission photomultiplier or the reflection mode.
- Laser adjustment with a transmission photomultiplier
- Use an empty well for adjustment.
- Choose line-scanning mode and histogram view.
- Decrease the laser intensity to the minimum, and adjust the detector gain to detectable background noise.
- Increase the laser intensity in steps of 0.5% and record the corresponding signal.
- Apply the routine for both laser lines.
- Laser adjustment with reflection mode
- Use an empty well for adjustment.
- Apply a reflection filter, switch on the reflection mode, if available.
- Ensure that the detector wavelength range covers the wavelength of the laser.
- Choose the line-scanning mode and histogram view.
- Decrease the laser intensity to the minimum, and adjust the detector gain to detectable background noise.
- Move the objective to the lowest position.
- Move the objective up until the reflection of the coverslip is visible.
- Increase the laser intensity in steps of 0.5% and record the corresponding signal.
- Apply the routine for both laser lines.
- Data evaluation
- Tabulate the data and sort the data by signal intensities.
- Plot the signal intensities against the relative laser power.
- Choose laser intensities that result in similar signal intensity.
3. Adjustment of photomultipliers
NOTE: After laser adjustment, the photomultipliers were adjusted to individual gains to obtain similar sensitivity. This calibration was done with the 514 nm laser line, which is in the center of the wavelength range of interest.
- Use an empty well for adjustment.
- Apply a reflection filter, and switch to reflection mode if available.
- Ensure that the detector wavelength range covers the wavelength of the laser (514 nm).
- Choose the line scanning mode and histogram view.
- Decrease detector gain to half the maximum, and adjust the laser intensity to detectable background noise.
- Move the objective to the lowest position.
- Move the objective up until the reflection of the coverslip is visible.
- Increase the detector gain in steps of 50 to 100 V and record the corresponding signal.
- Apply steps 3.1 to 3.8 for both detectors.
- Data evaluation
- Plot the intensity against the detector gain for each detector.
- Choose the individual detector gains to obtain similar sensitivity.
4. FRET image acquisition
NOTE: Start with the sample of interest for setting up image acquisition.
- Choose the appropriate filters/dichroic mirrors, e.g., a double dichroic mirror MBS 458/514 for the FRET-pair ECFP/EYFP. Use the same dichroic mirror for all channels to enable line-by-line scanning. Select a water immersion objective for the imaging of living cells. Choose 12 bit- or 16-bit scanning and moderate scanning speed.
- Define the detection range, preferably 470-510 nm for donor detection and 530-600 nm for acceptor/FRET detection in the case of ECFP/EYFP. When using a 445 nm or 440 nm diode laser, use 450 to 510 nm as the detection range. In the case of an acousto-optic beam splitter (AOBS), define donor detection in the range of 450 to 500 nm to prevent unwanted acceptor detection.
- Apply the detector setting according to 3.10.2.
- Apply the laser setting according to 2.3.2. Revise the laser intensity based on the obtained laser power table, if required. Ensure that the signal-to-noise ratio covers the entire dynamic range of the detectors (intensity ranging from 0 to 4095 for 12-bit scanning).
- Keep laser intensities and detector gains constant. Use the pinhole diameter for fine-tuning.
NOTE: Keep in mind that changes in the pinhole diameter affect spatial resolution. - Perform the measurements (take images of at least 20 cells).
5. Determination of crosstalk corrections
NOTE: Cells expressing only the donor or the acceptor are required to determine donor spectral bleed-through (DSBT) and acceptor spectral bleed-through (ASBT), respectively. Keep the same settings described in section 4.
- Perform FRET measurements with cells expressing the donor fluorophore.
- Perform FRET measurements with cells expressing the acceptor fluorophore.
6. Calibration of the measurements according to Beemiller et al.13
NOTE: Cells expressing a donor-acceptor fusion of known FRET efficiency are required. Here, an ECFP-5 aa-EYFP-fusion with a FRET efficiency of 0.46 has been used4. Keep the same settings described in section 4.
- Perform FRET measurements with cells expressing the donor-acceptor fusion
7. Data evaluation
- Obtain line profiles of the cells, ensuring that each profile contains no more than one cell. Save the profiles as text files.
- Import the text files into a spreadsheet using the text file import option in the Data section.
- Read out the maximum values by applying the Max function.
- List the obtained values in a table, have a column each for donor emission ID, FRET emission IF, acceptor emission IA, and at least four data sets: donor only, acceptor only, donor-acceptor fusion, and measurement.
NOTE: Excitation of the donor also results in direct excitation of the acceptor and causes ASBT that is described by the α value. - Calculate the ASBT α values with the acceptor-only dataset using equation (1).
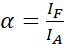 (1)
(1)
NOTE: Use the median of all α-values in the following equations. The donor shows a broad emission spectrum that results in emission crosstalk with the sensitized emission of the acceptor. This DSBT is given by the β value. - Calculate the donor spectral bleed-through β values with the donor-only dataset using equation (2).
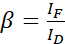 (2)
(2)
NOTE: Use the median of all β values in the following equations. The calibration factor ξ describes the linear relationship of FRET-derived donor quenching and sensitized emission of the acceptor. Use the medians of 7.5 and 7.6 in the following equations. - Calculate the calibration factors ξ with the donor-acceptor fusion dataset and its FRET efficiency E (0.46) using equation (3).
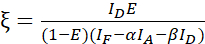 (3)
(3)
NOTE: Use the median of all ξ values in the following equations. - Calculate the FRET efficiencies of the protein pair of interest using equations (4) and (5).
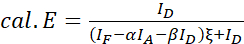 (4)
(4)
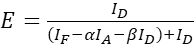 (5)
(5) - Estimate the effects of expression strength and/or donor-acceptor ratio: plot the sum of ID, IF, and IA against the FRET efficiencies. Perform a linear regression; note that the steeper the graph and the higher R2 is, the higher is the impact of the expression level or the greater is the difference of donor and acceptor abundance.
Adjustment of the confocal laser-scanning microscope
The laser adjustment revealed a linear increase of emission with increasing laser intensity (Figure 2 and Table 1). As expected for argon-ion lasers, the emission of the 514 nm line was much higher than the emission of the 458 nm line, as evidenced by a steeper slope. For subsequent experiments, laser power of 4.5% and 6.5% was chosen for the 514 nm line and the 458 nm line, respectively. This resulted in almost equal emission intensity of 1123 (514 nm) and 1141 (458 nm).
Varying the detector gains at constant laser power revealed an exponential behavior for both analyzed detectors. Similar emission intensities were obtained for a gain of 700 V (Figure 3). Although the adjustment of laser lines benefited from the linear behavior, the adjustment of the detectors is likely affected by the exponential behavior, resulting in marked differences with minor changes in the gain at higher voltages. Unfortunately, this higher range is of interest for measurements in living cells, as increasing the laser power is cytotoxic and promotes photobleaching.
Determining spectral bleed-through
At first, the spectral bleed-through of the donor and the acceptor were analyzed with recombinant purified ECFP and EYFP; DSBT β = 0.498 and ASBT α = 0.100. The same was done with cells expressing either ECFP or EYFP. With LSM 2, the estimated DSBT was β = 1.602 ± 0.207 (mean ± SD) and ASBT was α = 0.119 ± 0.018. The median values were β = 1.506 and α = 0.120. This discrepancy between the data obtained in living cells and the data obtained with recombinant protein demonstrates that it is impossible to omit the determination of spectral bleed-through in living cells. This is likely caused by cellular pigments.
The images reveal the higher brightness of EYFP in comparison to ECFP (Figure 4 and Table 2). Compensating the differences in brightness by different laser settings enhances the dynamic range of the donor and the FRET channel. Providing the relative output by measuring the laser intensities, as done for the adjustment, still increases the reproducibility of the measurements.
For LSM 1, β = 0.171 ± 0.044, α = 0.094 ± 0.031 with medians of α = 0.084 and β = 0.180. The determination of the ASBT depends on the laser adjustment and a single detector, and thus, the determination of ASBT performed well. The DBST involves two detectors, resulting in vastly different results for both microscopes. It should be remembered that LSM 2 is equipped with two photomultipliers, whereas LSM 1 uses a GaAsP detector and a photomultiplier, two different types of detectors with different spectral properties and sensitivity. Accordingly, the adjustment works better with two identical detectors.
Calibration of the measurement
For calibration, the median values of α and β were applied, and a fusion of ECFP and EYFP was used as the standard of known FRET efficiency (E = 0.46). The calibration of recombinant and purified ECFP-EYFP resulted in a ξ value of 13.44, while the in vivo measurements revealed a ξ value of 1.525 ± 1.844. The median was 0.798, revealing extreme outliers together with the high standard deviation. For LSM 1, the values were ξ = 1.978 ± 0.807 with a median of ξ = 1.883. For LSM 2 and LSM 1, the datasets obtained for the calculation of ξ (Table 3) were statistically identical, as proven by Student's t-test (p > 0.2).
FRET measurement
As a proof of concept, the FRET measurement was repeated with the donor-acceptor fusion. The measured FRET efficiency was 0.47 ± 0.07 for the purified protein and 0.47 ± 0.06 in living cells with LSM 2 (Table 4). The median of the measurements in living cells was E = 0.45. For LSM 1, the FRET efficiency was 0.46 ± 0.09 with a median of E = 0.45 (Table 4). These data demonstrate the effective calibration with a FRET construct of known FRET efficiency.
Effects of expression level and donor–acceptor ratio
For the analyzed data set, the FRET efficiencies were not influenced by the expression level. Due to the fusion of the donor and the acceptor, the ratio was also constant. The inclusion of previous datasets revealed a dependency on the expression level in one case (Figure 5). The FRET efficiency between the labeled vacuolar ATPase (V-ATPase) subunits, VHA-A-ECFP and VHA-a-EYFP, decreased with increasing signal intensity. In contrast, the interaction between VHA-E1-ECFP and VHA-C-EYFP was independent of the signal intensity. In the case of VHA-A and VHA-a, the three copies of VHA-A are increasingly replaced by VHA-A-ECFP, which results in a donor excess in comparison to the single copy of VHA-a in the complex. Although VHA-E1 might be present in the form of three copies, the high solubility of VHA-C might abolish this effect in this example. Here, the FRET efficiencies have been comparatively low. This simple approach allows for the testing of expression and ratio artifacts.
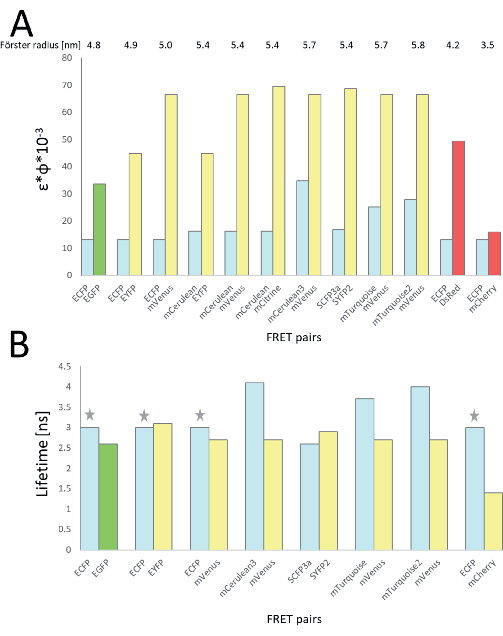
Figure 1: Overview of fluorescent protein FRET pairs with cyan-emitting donors. (A) The Förster radii of the pairs and the brightness of donor and acceptor are given for well-characterized FRET pairs. (B) Comparison of donor and acceptor lifetimes. Grey asterisks indicate a multiexponential decay. Abbreviations: FRET = Förster resonance energy transfer; CFP = cyan fluorescent protein; GFP = green fluorescent protein; YFP = yellow fluorescent protein; mX = monomeric X; EX = enhanced X; SX = super X. Please click here to view a larger version of this figure.
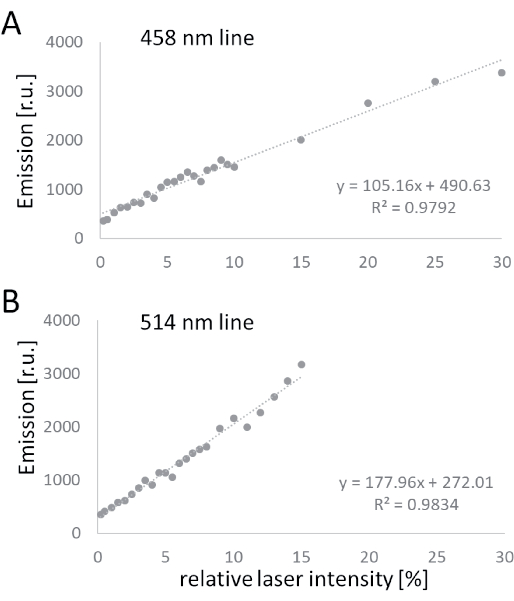
Figure 2: Laser adjustment. The emission of the lasers was recorded by the reflection of the coverslip and plotted against the relative laser intensity as modulated by the acousto-optic tunable filter of the LSM. Emission of the 458 nm line (A) and the 514 nm line (B) of an argon-ion laser is shown. Abbreviations: LSM = laser scanning microscope; r.u. = relative units. Please click here to view a larger version of this figure.
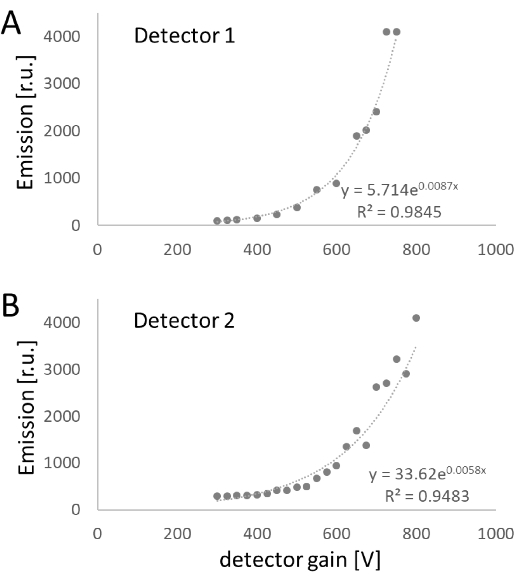
Figure 3: Detector gain and emission intensity. The resulting emission intensities were recorded in the reflection mode for detector gains ranging from 300 to 750 V and 300 to 750 V for detector 1 (A) and detector 2 (B), respectively. Abbreviation = r.u. = relative units. Please click here to view a larger version of this figure.
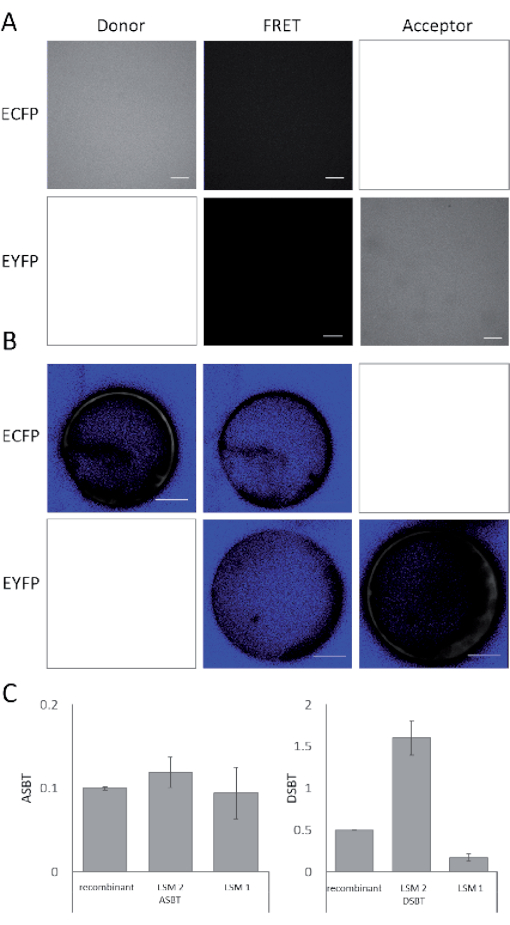
Figure 4: Spectral bleed-through. Images were obtained in the donor, the FRET, and the acceptor channels. (A) Images obtained with purified fluorescent proteins; scale bars = 100 µm; (B) corresponding images for cells expressing the fluorescent proteins; scale bars = 10 µm. (C) Graphs of the obtained SBT values; mean ± SD is given. Abbreviations: FRET = Förster resonance energy transfer; SBT = spectral bleed-through; ECFP = enhanced cyan fluorescent protein; EYFP = enhanced yellow fluorescent protein; LSM = laser scanning microscope; ASBT = acceptor SBT; DSBT = donor SBT. Please click here to view a larger version of this figure.
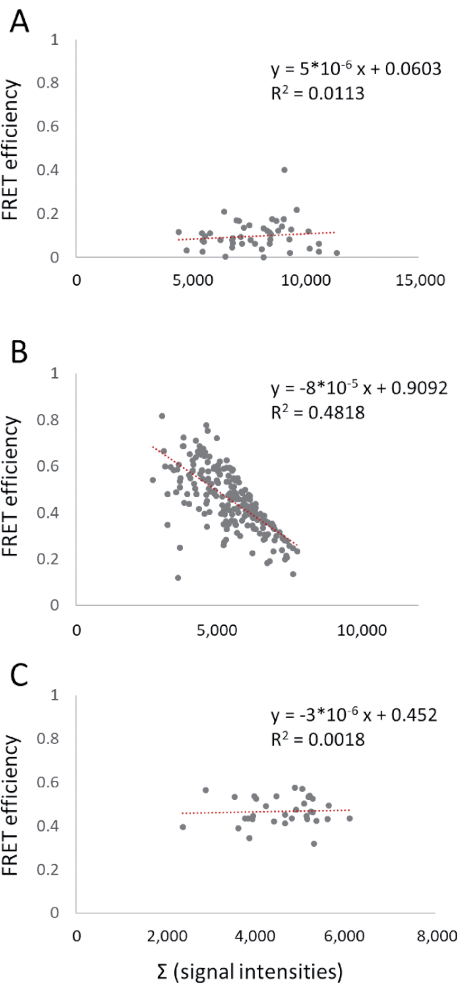
Figure 5: Effects of expression level and/or donor-acceptor ratio. The sum of the emissions in the donor, FRET, and acceptor channels has been plotted against the FRET efficiency. This has been done for VHA-E1-ECFP and VHA-C-EYFP (A), VHA-A-ECFP and VHA-a-EYFP (B), and the ECFP-EYFP fusion (C). Linear regression was performed; the equation and R2 are given. Abbreviations: FRET = Förster resonance energy transfer; ECFP = enhanced cyan fluorescent protein; EYFP = enhanced yellow fluorescent protein; VHA = vacuolar ATPase subunit. Please click here to view a larger version of this figure.
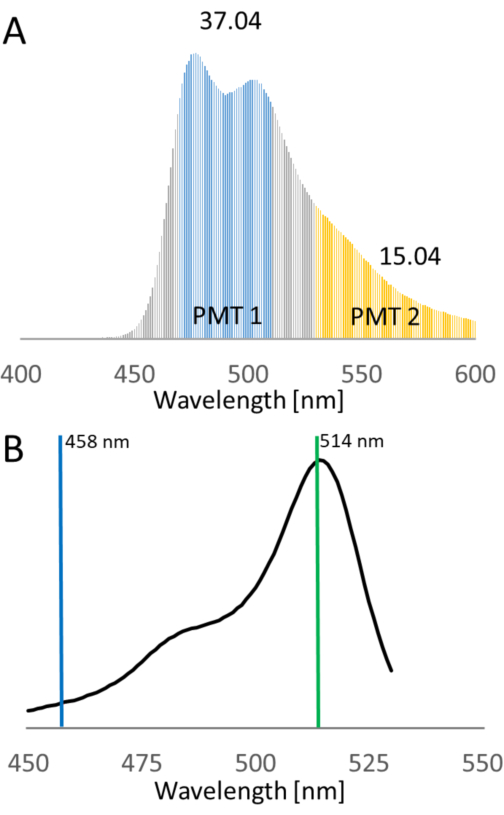
Figure 6: Donor and acceptor bleed-through of ECFP and EYFP. The spectra of the fluorescent proteins were obtained from the FP database14. (A) During sensitized emission experiments, the vast portions of the donor and the acceptor are detected by individual photomultipliers, named PMT1 and PMT2, respectively. ECFP emission is also detectable by the acceptor detector upon excitation with 458 nm. The calculated donor spectral bleed-through into PMT2 is 40.6% of the emission detected by PMT1. (B) The emission spectrum of EYFP demonstrates that EYFP shows direct excitation at 458 nm, which is frequently applied for the excitation of cyan donors. The expected excitation efficiency is 9.6% of the excitation at 514 nm. Abbreviations: FP = fluorescent protein; ECFP = enhanced cyan fluorescent protein; EYFP = enhanced yellow fluorescent protein; PMT = photomultiplier. Please click here to view a larger version of this figure.
Table 1: Laser power and signal intensity. The signal intensities of the 458 nm line are given in blue, the signal intensities of the 514 nm line in green. The intensities applied for the experiment are highlighted in grey. Please click here to download this Table.
Table 2: Signal intensities for SBT determination. Recorded maxima and calculated α and β values are given. Abbreviations: SBT = spectral bleed-through; LSM = laser scanning microscope; FRET = Förster resonance energy transfer; ASBT = acceptor SBT; DSBT = donor SBT. Please click here to download this Table.
Table 3: Signal intensities for calibration. Recorded maxima and calculated ξ values are given. Corrected values correspond to the FRET signal intensities minus SBT. Abbreviations: SBT = spectral bleed-through; LSM = laser scanning microscope; FRET = Förster resonance energy transfer. Please click here to download this Table.
Table 4: Signal intensities for FRET measurements. Recorded maxima and E values are given. Corrected values correspond to the FRET signal intensities minus SBT. Calibrated E values were calculated with calibration by ξ. Abbreviations: SBT = spectral bleed-through; LSM = laser scanning microscope; FRET = Förster resonance energy transfer; cal. = calibrated. Please click here to download this Table.
