Lineage Tracing of Inducible Fluorescently-Labeled Stem Cells in the Adult Mouse Brain
Summary
The ability to permanently mark stem cells and their progeny with a fluorophore using an inducible transgenic lineage tracing mouse line allows for spatial and temporal analysis of activation, proliferation, migration, and/or differentiation in vivo. Lineage tracing can reveal novel information about lineage commitment, response to intervention(s), and multipotency.
Abstract
A telomerase reverse transcriptase (Tert) lineage-tracing mouse line was developed to investigate the behavior and fate of adult tissue stem cells, by crossing the 'Tet-On' system oTet-Cre mouse with a novel reverse tetracycline transactivator (rtTA) transgene linked to the Tert promoter, which we have demonstrated marks a novel population of adult brain stem cells. Here, administration of the tetracycline derivative doxycycline to mTert-rtTA::oTet-Cre mice will indelibly mark a population of cells that express a 4.4 kb fragment of the promoter region of the gene Tert. When combined the Rosa-mTmG reporter, mTert-rtTA::oTet-Cre::Rosa-mTmG mice will express membrane tdTomato (mTomato) until doxycycline treatment induces the replacement of mTomato expression with membrane EGFP (mGFP) in cells that also express Tert. Therefore, when these triple-transgenic lineage tracing mice receive doxycycline (the "pulse" period during which TERT expressing cells are marked), these cells will become indelibly marked mGFP+ cells, which can be tracked for any desirable amount of time after doxycycline removal (the "chase" period), even if Tert expression is subsequently lost. Brains are then perfusion-fixed and processed for immunofluorescence and other downstream applications in order to interpret changes to stem cell activation, proliferation, lineage commitment, migration to various brain niches, and differentiation to mature cell types. Using this system, any rtTA mouse can be mated to oTet-Cre and a Rosa reporter to conduct doxycycline-inducible "pulse-chase" lineage tracing experiments using markers of stem cells.
Introduction
Value of a lineage tracing mouse line
Analysis of stem cells in vivo can be difficult since many assays that examine such cells only focus on characterizing these cells at the time of death of the animal, which represents a terminal snapshot in time. To better understand the processes of proliferation, differentiation, and migration of progenitor, intermediate/transition cell types, and mature cells over time, a longitudinal analysis approach is required. This can be achieved with lineage tracing studies whereby stem/progenitor cells are marked indelibly and can be followed for any length of time afterwards1.
In the adult mammalian brain, the process of neurogenesis by which adult-born neurons are created from stem and progenitor cells was first analyzed via label retention with thymidine-H3 2,3,4 or 5-bromo-2'-deoxyuridine (BrdU)5,6,7,8. In these studies, proliferative cells were marked with the thymine analog, which was incorporated into the cells' DNA during replication and cell division/proliferation. The marked cell, as well as their progeny, therefore contained this analog, which were then identified post-mortem. However, while thymidine-H3 and BrdU allowed for the marking of proliferative cells and their progeny in brain regions where stem cells were poorly understood, these studies were hindered by the downsides of these tools. Thymidine-H3 induces cell-cycle arrest, apoptosis, and dose-dependent DNA synthesis inhibition9, while BrdU marks cells at varying levels depending on the route of administration10 and is taken up by cells during repair or apoptosis, as well as during cell division11. More efficient labeling methods have been utilized recently, such as labeling with 5-ethynyl-2'-deoxyuridine (EdU), but many of the same issues as BrdU still remain12. These approaches are also limiting in that they mark any proliferative cell, not just stem cells, and thus interpretation of results can be confounded. In the neurogenic lineage, all stem and progenitor cells retain mitotic capacity, and only terminal/mature neurons are not proliferative. For astrocytes and microglia, but not oligodendrocytes, proliferation is maintained indefinitely13,14,15.
Transgenic mice used to lineage trace only cells that express a protein of interest, such as one confirmed to identify adult stem cells as we use here, have therefore become more common when investigating stem cells and their progeny. While difficult to generate through mouse transgenic approaches, lineage-tracing mouse lines allow for the tracing of specifically marked cells in the brain, and do not rely on proliferation alone. In the Tet-On transgenic mouse system, administration of tetracycline or doxycycline (a tetracycline derivative) induces Cre-recombinase expression in cells that have been engineered with a reverse tetracycline transactivator (rtTA), which is transcribed by a promoter of interest. The Tet-inducible Cre-driven recombination will then activate the expression of an indelible fluorescent or luminescent protein in the cells of interest, depending on the Rosa-reporter mouse employed. These indelibly marked cells continue to express this reporter after division, differentiation, or migration, allowing the tracking of these cells and their progeny over time or after different interventions16. Advantages of transgenic lineage-tracing approaches include: 1) specificity of tracing to a particular cell lineage or progenitor/stem cell marked by the rtTA, 2) indelible expression of the fluorescent or luminescent protein despite cell turnover or differentiation, 3) low toxicity, 4) conditional activation during any point in the animal's life cycle, and 5) ease of use with common assays, including immunostaining/immunofluorescence16.
Other methods of temporally inducible reporter tools in mice include the use of Cre-ERT2 mice, which can be paired with ROSA-GFP, ROSA-mTmG, or other fluorescent reporter transgenes. In these animals, a cell-specific regulatory element, such as a promoter or enhancer region of interest, drives the production of Cre recombinase, which can only be activated via the administration of tamoxifen. While Cre-ERT2 mouse lines allow for the induction of Cre in specific cell lines, there is a wealth of knowledge detailing the effects of tamoxifen on adult neurogenesis17,18. Additionally, there exists many ROSA-driven fluorescent reporter genes that can be utilized instead of GFP or mTmG, including YFP or CFP, which would allow for alternate fluorescent labeling in other fluorescent wavelengths. These fluorescent reporter genes can be used with Tet-On or Cre-ERT2 systems.
Telomerase reverse transcriptase (TERT) is the rate-limiting component of the holoenzyme telomerase, which works to extend telomeres after they are shortened during cell division19,20. TERT has been identified as a marker of adult tissue stem cells in the intestine21,22, bone marrow21, liver23, adipose24, endometrium25,26, and bone1. Whether TERT expression in these adult stem cells is solely for telomerase-extending activity or to perform non-canonical TERT roles27 is still unknown. TERT-expressing quiescent adult stem cells (qASCs) have been identified and traced throughout the body with transgenic lineage tracing mice to study the multipotency and self-renewal ability of these stem cells, as well as their potential to activate, proliferate, differentiate, and migrate1,22,23,28. The creation of the Tert-rtTA transgene has been performed previously1. In this paper, we will describe the use of a lineage tracing TERT mouse line to study TERT + qASCs which we have identified as a novel ASC population in the adult mouse brain.
Generation of a transgenic lineage tracing mouse line
To generate a lineage-tracing mouse line using the Tet-On system, three transgenes must be combined within a single animal through mouse mating. The first is a rtTA, expressed under the control of the promoter of the gene of interest (ours is TERT-rtTA). In a cell that expresses this gene of interest, the rtTA will therefore be expressed. The second is an oTet-Cre gene, which contains a tetracycline response element (TRE) that will allow for the transcription of Cre recombinase in the presence of both a rtTA fusion transcript and tetracycline or doxycycline. Tetracycline or doxycycline can be administered to an animal via drinking water or chow29. Finally, there must be a gene that will be activated by Cre recombinase cleavage. In this manuscript, the labeling gene we will describe is the Rosa26-mTmG gene, which until Cre recombination will ubiquitously transcribe mTomato (membrane red fluorescence in all cells). However, if a cell expresses the rtTA transcript and contains tetracycline or doxycycline, Cre recombination of a set of lox P sites between the mTomato and mGFP sites will change the R26 site and cause the cell to produce membrane EGFP (mGFP, or membrane green fluorescence) instead of membrane tomato. The indelible nature of this mGFP signal will allow for in vivo labeling and tracing of cells as they proliferate, migrate, and differentiate. Following the trace, cells can be analyzed for expression of GFP via immunofluorescence in standard cryosections or thicker optically cleared brains.
In this paper, we will describe the use of the specific mouse line, mTert-rtTA::oTet-Cre::Rosa-mTmG. To create this triple transgenic mouse line, we first mated oTet-Cre animals (The Jackson Lab strain #006234) to Rosa-mTmG animals (The Jackson Lab strain #007676) to create oTet-Cre::Rosa-mTmG double transgenic mice. These animals were genotyped with primers and PCR templates indicated in Supplementary Tables 1 and 2. mTert-rtTA mice (created by David Breault1) were mated to oTet-Cre::Rosa-mTmG animals to create mTert-rtTA::oTet-Cre::Rosa-mTmG mice (Figure 1A)1,30. The mechanism of action of the mTert-rtTA::oTet-Cre::Rosa-mTmG mouse line is illustrated in Figure 1B.
Doxycycline induction and pulse-chase design
When planning experiments, it is important to consider several factors that will affect the outcome of lineage tracing experiments, including the age of the animal at doxycycline induction, the length of doxycycline administration (the "pulse" period), the length of time after doxycycline removal before tissue collection (the "chase" period), and the timing of any interventions during these processes. These study design considerations are essential for understanding the labeled cells and their progeny at the conclusion of the experiment. The first step in the process is to identify the age of interest of the animal and the length of the doxycycline administration. Longer pulse periods will allow for the potential for more cells to express the rtTA-linked promoter to be expressed and more cells of interest to be indelibly marked. A cell type that exhibits transient expression of the gene of interest may require a longer pulse period than cells that continuously express the gene of interest. However, if the goal of the study is to understand short-term effects of the cell of interest or an acute intervention, the pulse period cannot be too long, as once a cell is marked, it will be traced through any number of changes during the remainder of the pulse period. In some of our studies, a 2 day pulse with no chase period was used to more closely mimic a direct-reporter for TERT. This is because the minimum length of time for recombination of the Rosa-mTmG gene is 2 days31.
The next essential period in a pulse-chase experiment is the length between removal of doxycycline and perfusion of the animal, also known as the 'chase'. The half-life of doxycycline in mice is approximately 170 minutes regardless of administration route, allowing for the conclusion that doxycycline induction is likely no longer occurring several hours after removal32. However, it is important to note that doxycycline metabolism is slower in aged mice, which may lead to longer effective 'pulse' periods than young animals33.
During the chase period, labeled cells will continue to be labeled, even after proliferation, differentiation, or migration. The progeny of these cells will also be labeled. The goal of the study will shape the length of the pulse-chase paradigm. If the goal of the study is to understand the regeneration of a tissue over a long period of time with the cells of interest, a long chase period may be required. Finally, the timing of any interventions or treatments must be decided. Administration during the pulse period will affect the cells expressing the gene of interest as well as any progeny created during this time, whereas administration during the chase period may affect mostly progeny of the cells of interest, although this will depend on how quickly the labeled cells divide or differentiate. Examples of pulse-chase experimental designs we have utilized are outlined in Figure 2A.
It is also important to be aware of the possibility of background GFP signal due to leaky expression. Leaky expression occurs as a result of inherent binding potential between rtTA and the Otet sequences in the absence of doxycycline, and residual activity of the Otet transgene in the absence of rtTA (reviewed in34). Leaky expression represents one weakness of the Tet systems, and although the leak is often an acceptable downside to the system, situations where the leaky expression can lead to toxicity and mortality, such as with diptheria toxin (DTA) in Otet-DTA animals can limit the use of these systems35.
Brain processing, sectioning, and immunofluorescence of thin sections for microscopy
To analyze the brains of mice after a lineage tracing experiment, mice must first be perfused via transcardial perfusion to remove blood and CSF that can lead to high autofluorescence in the brain. There are two commonly used fixatives that the animal may be perfused with: Histochoice Tissue Fixative, a glyoxal fixative (GF), or 4% paraformaldehyde (PFA). Glyoxal fixatives allow for a relatively gentle fixation process with less cross-linking than PFA. This reduces tissue stiffness, reduces the need for antigen retrieval, and allows for less concentrated antibody solutions during immunostaining. However, if they contain methanol this may serve to increase autofluorescence36. On the other hand, PFA will often allow for a more robust fixation, and while antigen retrieval will be required during immunostaining, there are antibodies that will only work with PFA-fixed tissues, and perfusion with 4% PFA is required for the optical clearing technique described later.
Following perfusion is a post-fixation step, in which brains are immersed in the same type of fixative utilized during the perfusion overnight. This allows for any brain regions that may not have been fully fixed during the perfusion to continue to fix. Next, brains will be incubated in sucrose in phosphate buffered saline (PBS) to remove as much water as possible before the freezing step to prevent ice formation within the tissue ("cryo-preservation"). Brains may then be cut into any number of sagittal, coronal, or transverse tissue sections for processing. For both precision and reproducibility, calibrated brain blocks need to be used in a way that ensures consistent bregma cuts between animals. The most important factor that can affect how brains are sectioned is the brain region(s) of interest. For example, while a sagittal cut may allow for more brain regions to be analyzed in one tissue section, this cut will not allow analysis of neuro/gliogenic regions of the ventricular-subventricular zone (V-SVZ) as the lateral and medial sides of the lateral ventricle will be impossible to discern in that dimension and are better observed in the coronal plane. This can be an important distinction to make in adult neurogenesis studies, as the lateral side of the lateral ventricle contains the neurogenic V-SVZ, while the medial wall of the lateral ventricle is a more gliogenic niche37.
After tissues have been divided into coronal, sagittal, or transverse sections, they will be frozen into blocks using Optimal Cooling Temperature (OCT) embedding solution. Here, brains are frozen within this embedding material, with the use of a mixture of dry ice and ethanol. If thin section analysis is required, blocks will be sliced on a cryostat, and depending on the downstream analysis required can be adhered to positively charged glass slides as thin sections (<20 µm). Glass slides that are not charged can also be used, but the use of uncharged slides may result in tissues becoming unstuck from the slides38. If medium-thickness free-floating tissue sections (>20 µm) are required, tissues will be collected as free-floating sections. If thick sections (0.5-4 mm) are required, slicing non-frozen brain sections with a vibratome is required. It is important to note the storage differences between sections on slides, which require freezing at -20 to -80 °C and free-floating sections, which will be stored at 4 °C.
Brain clearing and whole mount immunofluorescent confocal microscopy
If a broader, yet less detailed analysis of the lineage-traced cells in the mouse brain is desired, a comprehensive analysis of thicker segments of brain tissue is possible with iDISCO brain clearing39 followed by immunostaining and tiled z-stack confocal microscopy or light-sheet microscopy. Here, large sections of mouse brain will be optically cleared (a process of delipidation39), which better allows for fluorescent signal imaging across a 1 mm tissue section. The downsides of this process are high autofluorescence and a diminished ability to obtain high resolution images of cell morphology and detailed co-staining analysis due to the increased working distances required when compared to more traditional methods (thin cryosections or free-floating sections). For this reason, we recommend brain clearing to analyze the large-scale lineage tracing results throughout multiple brain areas, instead of attempting to characterize or analyze individual cells.
Protocol
All methods described here have been approved by the Institutional Animal Care and Use Committee (IACUC) of The Ohio State University.
1. Creation of a lineage tracing Tet-On mouse line, breeding strategies, and genotyping for experimental cohort mice
NOTE: Here, we outline creation of a Tet-On mouse system with Rosa-mTmG as the fluorescent reporter gene that undergoes Cre recombination, however various other Cre-recombination-driven reporter lines can be utilized with the Tet-On system as well.
- Set up a mating cage of one oTet-Cre (+/-) male with one or more R26 (mTmG) (+/+) females. See Figure 1 for an overview of the experimental mouse mating setup.
- Resulting pups (F1) will be oTet-Cre (+/-) or oTet-Cre (-/-) and R26 (mTmG) (+/-). Perform genotyping according to the primers in Supplementary Table 1 and protocols in Supplementary Table 2.
- At weaning, perform germline testing by taking an ear punch from each weanling. Examine the ear punch under an epifluorescent microscope in the GFP/FITC spectrum to determine whether the animal had undergone germline recombination in development. If so, the animal will express GFP in every cell, and the ear punch will fluoresce with mGFP signal. These animals cannot be used for matings or experiments.
NOTE: Ear clips from mice that are not germline show only endogenous fluorescence under the microscope (Figure 1C), while germline animals show a bright GFP signal (Figure 1D). Control ears can be from mice that do not contain fluorescent transgenes. As a note, ear hairs have high endogenous fluorescence and must not be used as a determining factor (Figure 1C). Additionally, differences in endogenous fluorescence are observed between mice of white vs. black coat color, and controls from mice of the same coat color as mice being tested will need to be used in this analysis. - Mate one male oTet-Cre (+/-) x R26 (mTmG) (+/-) with one or more female oTet-Cre (-/-) x R26 (mTmG) mice.
- The resulting generation (F2) will be ¼ R26 (mTmG) (+/+) and ½ Cre (+/-). Cross these animals with mTert-rtTA (+/+) animals by mating one oTet-Cre (+/-) x R26 (mTmG) (+/+) with one or more mTert-rtTA (+/+) animals.
NOTE: We used mTert-rtTA (+/+) animals, but this process can be performed with any rtTA mouse line30. - For experimental animals, set up breedings to generate mTert-rtTA (+/+) or (+/-) x oTet-Cre (+/-) x R26 (mTmG) (+/+) or (+/-).
2. Doxycycline induction of Cre and pulse-chase design
- When animals are at the correct age for the study to begin, replace their drinking water with dox water: drinking water containing 5% sucrose and 2 mg/mL doxycycline hyclate. Use Cre-negative animals that receive the same doxycycline pulse-chase as experimental animals as a control group for the resulting fluorescent expression patterns.
- After preparation, store dox water at 4 °C for up to 1 month. See Table 1 for more information. Dox water may be administered to animals in drinking bottles for up to 5 days before being changed out, as fungal growth can occur if dox water is left out at room temperature for longer periods of time.
NOTE: A doxycycline pulse of at least 2 days is required for induction of mGFP signal when utilizing the mTmG transgene (initial mTmG paper).
NOTE: We have not tested doxycycline concentrations other than 2 mg/mL. Previous literature has identified that this concentration has the highest effects when administered through drinking water29. The amount of doxycycline delivered may differ depending on the amount of drinking done by each animal. Doxycycline injection or gavage may also be done in order to be consistent between animals40.
- After preparation, store dox water at 4 °C for up to 1 month. See Table 1 for more information. Dox water may be administered to animals in drinking bottles for up to 5 days before being changed out, as fungal growth can occur if dox water is left out at room temperature for longer periods of time.
- After the pulse period has ended, remove dox water from the cages and replace with normal drinking water. The animals will now be in the 'chase' period until death.
3. Transcardial perfusion and brain dissection
- Remove a mouse from its cage and restrain the animal using the left thumb and forefinger. Inject the animal via intraperitoneal (i.p.) injection with a solution of 100 mg/mL ketamine and 20 mg/mL xylazine in 0.9% sterile saline for a final concentration of 200 mg/kg ketamine and 20 mg/kg xylazine. See Table 1 for more information.
CAUTION: The protocols for anesthesia may differ depending on the country. Additionally, there are effects of anesthetics on the brain including changes in microglial morphology and action and corticosterone levels that must be understood when performing perfusions for the purpose of brain collection and analysis41,42. - After approximately 1-2 min the animal will lose its righting reflex. To test this, simply roll the animal on its back and if it cannot roll back onto its feet, it has lost the righting reflex.
- After approximately 5-10 min the animal will lose its pedal reflex. To check the pedal reflex, use the thumb and forefinger to pinch each of the animal's feet. If there is no response, in the form of a jump or muscle spasm, use the nails of the thumb and forefinger to pinch each of the animal's feet. If there is no response, the animal has lost the pedal reflex.
NOTE: If the animal responds to the pedal reflex for greater than 15 min after injection, an additional injection of ½ of the initial ketamine/xylazine injection may be required. Age, sex, genotype, phenotype, and treatment may affect the response of the animal to this drug cocktail. - After the righting and pedal reflexes are lost, test the ocular reflex by lightly contacting the mouse eyeball with a gloved finger. A lack of response indicates that the ocular reflex has been lost.
- Secure the mouse in a supine position with paws pinned to a pinnable work surface.
- Make an incision through the skin with surgical scissors along the thoracic midline approximately 1-2 cm below the xiphoid process. Cut superior until at the xiphoid process.
- Grasp the cartilage of the xiphoid process with forceps. Insert scissors and cut through the musculature and ribcage diagonally along the mouse's right side to the level of the clavicles.
- Raise the thoracic musculature with forceps and slice through the diaphragm until the heart can be visualized. Then make an identical diagonal cut along the mouse's left side, making sure to prevent any contact with the heart. Use a hemostat to keep the thoracic musculature away from the cavity.
NOTE: At this point, the mouse will not be able to breathe any longer, so work quickly to prevent death prior to the next steps.
- Raise the thoracic musculature with forceps and slice through the diaphragm until the heart can be visualized. Then make an identical diagonal cut along the mouse's left side, making sure to prevent any contact with the heart. Use a hemostat to keep the thoracic musculature away from the cavity.
- Secure the beating heart with blunt forceps and insert a dispensing needle into the left ventricle through the base of the aortic arch.
- Clamp the needle base to the left ventricle just above the insertion site.
- Make an incision in the right atrium, and at the first sign of blood flow, begin perfusing the animal with 20 mL of 1x PBS at 8.11 mL/minute.
- After the 1x PBS has perfused the body, switch to the appropriate fixative for the downstream application and perfuse the mouse with 20 mL of fixative.
- For frozen sectioning and immunostaining, perfuse animal with the glyoxal fixative for brain clearing, perfuse animal with 4% PFA in 1x PBS (pH 7.4).
NOTE: Signs of a successful perfusion include animal stiffness (slight stiffness with glyoxal, intense stiffness with PFA) and a lack of visible blood vessels throughout the tissues.
- For frozen sectioning and immunostaining, perfuse animal with the glyoxal fixative for brain clearing, perfuse animal with 4% PFA in 1x PBS (pH 7.4).
- Decapitate the mouse and remove the brain by inserting scissors into the brainstem and cutting along the squamosal suture through to the frontomaxillary suture on each side. Then cut across the fontonasal suture to remove the skull cap, being careful not to damage the olfactory bulb. From there, turn the skull upside down, and use curved forceps to remove the brain, making sure to sever the optic nerve.
- Place the perfused brain in a tissue cartridge and immerse in the fixative used to perfuse the animal overnight at 4 °C.
4. Brain treatment, slicing, and immunostaining
- Brain treatment and slicing
- Remove brain cartridges from the glyoxal fixative and incubate in 15% sucrose in 1x PBS for 2 days at 4 °C or until brains sink.
- Remove brain cartridges from 15% sucrose and incubate in 30% sucrose in 1x PBS for 2 days at 4 °C or until brains sink.
NOTE: See Table 1 for more information. - Remove brains from 30% sucrose in 1x PBS and separate brain into various 'blocks' using a coronal brain block or sagittal brain block.
- Embed coronal or sagittal brain sections in OCT compound by placing brain sections on a mixture of dry ice and ethanol (EtOH). After OCT has turned white and become solid, remove from dry ice-EtOH mixture and store wrapped in parafilm, inside an air-tight bag at -20 °C.
- Remove frozen blocks from the freezer and place inside a cryostat with an internal temperature set to -20 °C. Use OCT to attach the block to a metal chuck so that the tissue is solidly attached. Allow ~5 min for the tissue to freeze to the chuck. Slice the tissue at 7 µm, placing each tissue slice on a charged glass slide.
- Allow for slides to bake overnight at 37 °C, and then place at -20 °C until used for immunostaining.
- Immunostaining
- Warm slides to room temperature (RT) and post-fix with ice-cold acetone for 15 min.
- Wash slides for 5 min in 1x rinse buffer (e.g., IHC Select TBS Rinse Buffer), shaking at 60 rpm at RT between each step.
- Permeabilize for staining of nuclear antigens with 0.3% Triton X-100 for 10 min at RT. Permeabilize for staining of cytoplasmic antigens with 0.3% Tween-20 for 10 min at RT.
- Perform antigen retrieval by microwaving slides in 50-100 mL of 1x DAKO Antigen Retrieval Solution on low for 10 min, twice. Then rinse with rinse buffer for 5 min at RT.
- Incubate slides in 0.3% Typogen Black in 70% EtOH for 20 min at RT and then wash with rinse buffer.
- Draw a hydrophobic barrier around each tissue without allowing tissues to dry. Then block for 20 minutes at 37 °C with 1 drop of Millipore Blocking Reagent per tissue. Then add 100 μL primary antibody diluted in antibody diluent to each tissue and incubate overnight at 4 °C.
- The next day, incubate sections in Alexa Fluor secondary antibodies for 10 min at RT. Then wash slides in rinse buffer.
- Cover brain sections in 100 μL of 1:500 anti-GFP antibody conjugated to AlexaFluor 488 to boost the endogenous GFP signal marking traced cells and incubate overnight at 4 °C.
- The next day, wash with rinse buffer 2x, and then wash in running DI water for 5 min.
NOTE: Be sure to gently wash with DI water, taking care to avoid any running water on slides themselves that could remove brain sections from slides. - Counterstain with 100 ng/mL 4′,6-diamidino-2-phenylindole (DAPI) for 5 min. Then wash in running DI water for 5 min.
- Add a drop of mounting medium on top of each tissue section and seal with a 22 mm x 22 mm coverslip. Imaging is recommended on the day of mounting to ensure optimal signal.
5. iDISCO brain clearing (adapted from39)
- Fixation and sectioning
- Post-fix brains in 4% PFA overnight at 4 °C and then again for 1 h at RT.
- Wash brains in 1x PBS for an hour, twice, at RT. Then cut into 1 mm thick sections using the necessary brain block and place into 5 mL microcentrifuge tubes containing 1x PBS.
- Methanol pre-treatment (with methanol)
NOTE: Due to the possibility of methanol impacting immunostaining of certain proteins, the authors would like to point the reader to Renier et al. 2014 for a protocol to a methanol free iDISCO protocol alternative39. See Table 1 for more information regarding the following solutions in this section.- Wash with 1x PBS for 1 h twice (shaking) at RT.
- Wash in 50% methanol (in PBS) for 1 h (shaking) at RT.
- Wash in 80% methanol for 1 h (shaking) at RT.
- Wash in 100% methanol for 1 h twice (shaking) at RT.
- Bleach samples with 5% hydrogen peroxide (H2O2) in 20% dimethyl sulfoxide (DMSO)/methanol (1 volume 30% H2O2/1 volume DMSO/4 volume methanol, ice cold) at 4 °C overnight (shaking).
- After bleaching, wash samples in methanol for 1 h twice (shaking) at RT.
- Wash in 20% DMSO/methanol for 1 h twice (shaking) at RT.
- Wash in 80% methanol for 1 h (shaking) at RT.
- Wash in 50% methanol for 1 h (shaking) at RT.
- Wash in PBS for 1 h twice (shaking) at RT.
- Wash in PBS/0.2% Triton X-100 for 1 h twice (shaking) at RT.
- Immunostaining of clearing brains
- Incubate samples in 1x PBS/0.2% Triton X-100/20% DMSO/0.3 M glycine at 37 °C overnight on an orbital shaker.
- Block in 1x PBS/0.2% Triton X-100/10% DMSO/6% goat serum at 37 °C for 3 days on an orbital shaker.
- Wash samples in 1x PBS/0.2% Tween-20/10 µg/mL heparin sodium salt from porcine mucosa for 1 h twice at 37 °C, and then incubate in 1x PBS/0.2% Tween-20/10 µg/mL heparin/5% DMSO/3% goat serum containing 1:500 concentration of rabbit anti-GFP AlexaFluor 488 at 37 °C on an orbital shaker for 2 days.
- Wash samples in 1x PBS/0.2% Tween-20 with 10 µg/mL heparin for 1 h at 37 °C on orbital shaker, three times, and then once a day for 2 days.
- Incubate samples overnight in 10 mL of 50% v/v Tetrahydrofuran (THF)/H2O in a 15 mL conical tube.
- Incubate samples for 1 h in 10 mL of 80% THF/H2O.
- Incubate samples twice for 1 h in 100% THF.
- Dry samples with a sterile wipe and incubate in dichloromethane until they sink to the bottom of the vial (approximately 40 min). Do not incubate >60 min.
NOTE: Be sure to handle dichloromethane under a fume hood. - Incubate samples in 18 mL of dibenzyl ether (DBE) until clear (>2 h).
- Store samples in DBE at RT until ready to image.
NOTE: For best results, it is important to image samples as soon as possible after clearing is complete.
6. Imaging of stained slides or cleared brains
- To image immunostained brain sections, analyze slides via confocal microscopy, where co-expression of multiple antibodies can be confirmed. We use a Leica Stellaris 5 microscope with HyD S hybrid detectors, but any point scanning confocal microscope will work. The objectives include HC PL APO 10x/0.40 CS2, HC PL APO 40x/1.30 OIL CS2, and HC PL APO 63x/1.40.
- Configure the correct laser lines and emission filters. The required lasers and corresponding laser intensities are subject to the microscope and fluorophore combinations being used.
NOTE: We utilize a combination of diode 405 nm and white light lasers to fine tune excitation and emission spectra for each fluorophore, or groups of fluorophores, being used to reduce and eliminate crosstalk. Similar outcomes can be achieved using traditional laser/filter combinations as well but pay close attention to channel bleed through. Bleed through can be identified by turning on a single laser line in conjunction with each of the desired emission filter combinations to see what channels are impacted by each specific laser. For example, if the diode 405 nm laser is producing visible fluorescence in the GFP emission filter, there is bleed through occurring. Ensure that multiple channels be imaged sequentially frame by frame to greatly reduce any bleed through from occurring. - For co-stained Tet-On brains with an mTmG reporter, select DAPI (ex. 405 nm, em. 450 nm), GFP (ex. 488 nm, em. 509 nm), tdTomato (ex. 555 nm, em. 582 nm), and the corresponding far-red fluorophore to match the fluorophore used on the secondary antibody. We recommend Alexa Fluor 647 (ex. 651 nm, em. 667 nm).
- Establish settings first with a Cre- control brain stained with both GFP and the fluorescent secondary. These brain slices will control for background fluorescence from the fluorescent antibodies and will show no real GFP signal.
- Analyze each brain section from top to bottom, left to right, to verify that no signal has been missed in the analysis. If imaging the same brain section at various magnifications, it is recommended that the lower magnifications are utilized first, as the higher magnification will more quickly bleach the mTomato signal.
- Configure the correct laser lines and emission filters. The required lasers and corresponding laser intensities are subject to the microscope and fluorophore combinations being used.
- To image cleared brains, use an inverted confocal microscope with an automated stage and automated tiling function. We use the Leica Stellaris 5 microscope. Because the samples are so thick (>500 µm), larger working distances are required which limits the magnification that can be effectively achieved. We use the HC PL APO 10x/0.40 CS2 objective for all cleared brain imaging.
- Start by laying a cleared brain section flat against a glass bottom dish and submerge in dibenzyl ether.
NOTE: Dibenzyl ether is corrosive to most objective lenses and most plastics. For this reason, any interior plastic in the glass bottom dish needs to be coated with quick drying silicone elastomer. - Configure the correct laser lines and emission filters. For co-stained Tet-On brains with an mTmG reporter, select DAPI, GFP, tdTomato, and the corresponding far-red fluorophore to match the fluorophore used on the secondary antibody. Set the desired resolution, we recommend at least 1024 x 1024. Set pinhole to 1 AU.
- Establish the laser intensity and detector gain first with a Cre- control brain stained with both GFP and the fluorescent secondary. These brain slices will control for background fluorescence from the fluorescent antibodies and will show no real GFP signal.
- Once laser intensities and detector gain have been optimized, map out the brain for imaging. Start by locating the entire perimeter of the brain section to set the X and Y axis of the image acquisition. Next, identify the Z axis by setting the focal point to approximately the tissue's center depth, and use Z-positioning controls to locate the "highest" point of the tissue (most positive Z-value). Scan various regions of the tissue increasing the maximum Z-axis height as necessary.
- Repeat for the "lowest" (most negative Z-value) point required to obtain the entire thickness of the tissue. The Stellaris 5 has a limit of ± 250 um of the focal point. This limits optical sections to 500 µm thick in the Z-axis. The 3D area of the brain section is now known.
- Set Z-step size to 6 µm and start acquiring the image. Acquisition time is dependent on several factors and can range from hours to days depending on the desired parameters. To decrease image acquisition time try decreasing resolutions, increasing laser scan speed, or increasing step size.
- Once the tiled image of the entire cleared brain section has been captured, analyze as desired.
- Start by laying a cleared brain section flat against a glass bottom dish and submerge in dibenzyl ether.
Representative Results
While the fluorescent signal resulting from a Tet-On system with a fluorescent reporter will vary depending on the promoter of interest and fluorophore(s) used, we will describe how findings were analyzed with mTert-rtTA::oTet-Cre::Rosa-mTmG animals. Analysis of adult brains after a pulse-chase resulted in membrane GFP-expressing cells throughout various anatomical niches. The difference in fluorescent intensity between various cell types must be noted. One variable regarding fluorescent intensity is the size of the cell membrane. For instance, choroid epithelial cells (CPECs) in the choroid plexus have a thick cell membrane and strong fluorescent signal that was easily distinguished from the surrounding cells (Figure 4A,B). However, other smaller and currently unidentified cell types in the choroid plexus stroma had a weaker GFP signal (Figure 4C). In the cerebellum, Bergmann glial cells expressed higher levels of mGFP than basket cells, and variation in mGFP expression even existed between individual basket cells (Figure 4D). Olfactory sensory neuron axons within the glomerular layer of the olfactory bulb also expressed high levels of mGFP (Figure 4A,E). To ensure that all mGFP+ cells are identified, it is therefore important to identify both bright and dim GFP+ cells throughout the brains of mTmG animals. In mTert-rtTA::oTet-Cre::Rosa-mTmG animals, we observed low background mGFP expression across brain regions including the cerebral cortex (Figure 4A). Interestingly, the cerebellum and brainstem showed lower background mGFP expression in this manner (Figure 4A).
During analysis of brain tissue after a lineage trace there will be differences in mGFP and mTomato expression patterns across both brain regions and cell types. Olfactory sensory neuron axons that fill the glomerular layer of the olfactory bulb expressed high levels of membrane tomato when compared to most other brain regions, even when the same neurons are mGFP+, indicating that some cells appeared to lose mTomato signal more slowly (Figure 4D). In contrast, in the choroid plexus, mGFP+ cells expressed low mTomato (Figure 4B,C). In most other brain regions tomato signal is spread evenly between cell types, with endothelial cells being some of the only cells whose fluorescent signal was bright enough to stand out from the red fluorescent background (Figure 4B). The activation of mGFP expression and cleavage of mTomato from the genome during recombination results in a loss of mTomato expression, but the mTomato fluorophores that are expressed on the membrane are retained until they degrade. For this reason, mGFP+ cells will often express variable mTomato signal, which will depend on the recency of the recombination, the cell type, and size of the cell membrane. Therefore, it is important to analyze cells based on the expression of mGFP and to not exclude them from analysis if they express both mGFP and mTomato.
Alternative reporters may also be utilized in conjunction with the Tet-On system. Cre recombination may act to induce expression of GFP or RFP in cells that will normally not express any fluorescent tag in Rosa-GFP or Rosa-RFP animals. Here, analysis of GFP or RFP will indicate the expression of the promoter of interest, and no fluorophore signal will be removed such as with mTmG animals. Single-fluorophore reporters allow for immunostaining with more fluorescent antibody combinations, as the membrane tomato in mTmG animals prevents staining in the red wavelength.
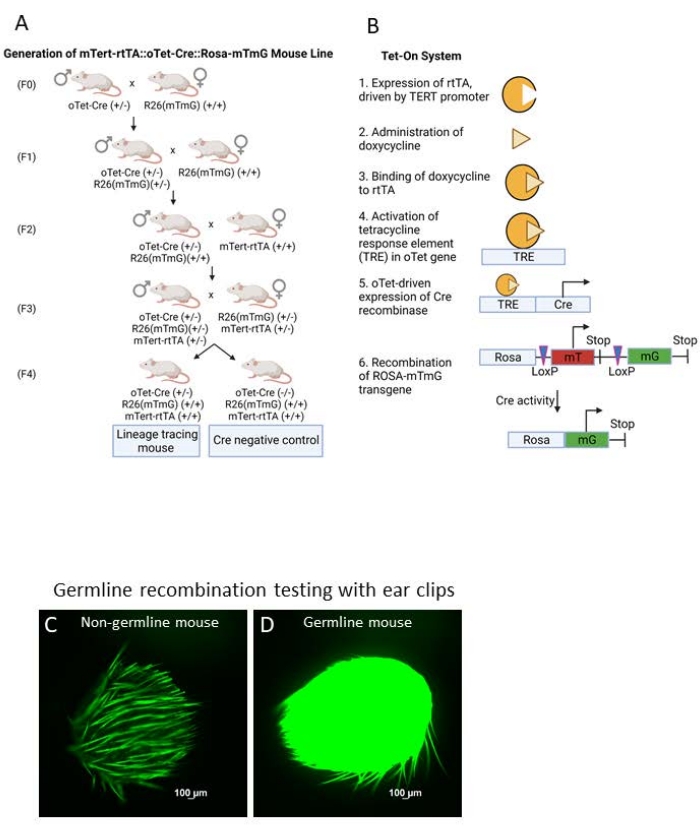
Figure 1. Generation of a Tet-On mouse line. (A) Outline of the breeding scheme for the creation of mTert-rtTA::oTet-Cre::Rosa-mTmG mice from individual mouse lines. (B) Schematic of the Tet-On system in the mTert-rtTA::oTet-Cre::Rosa-mTmG mouse line. (C–D) Representative images of ear clips imaged with FITC channel of an epifluorescent microscope from non-germline (C) and germline (D) mTert-rtTA::oTet-Cre::Rosa-mTmG mice. Please click here to view a larger version of this figure.
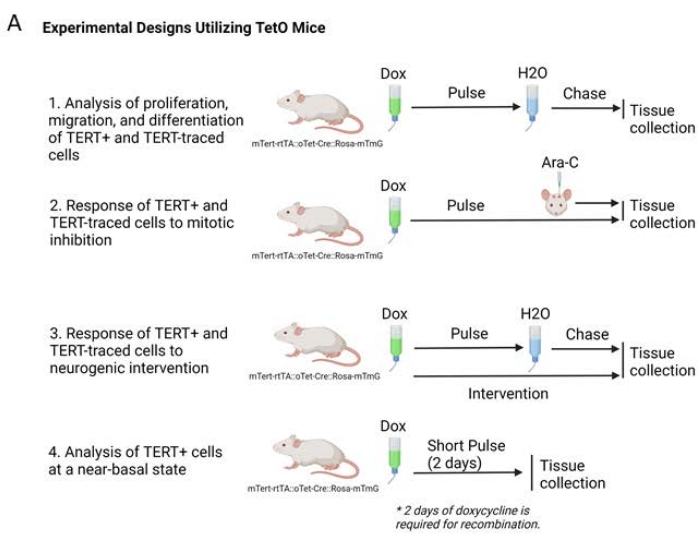
Figure 2. Experimental designs with lineage tracing Tet-On mice. Illustrations of experiments possible with Tet-On mice from basal proliferation, migration, and differentiation (1) to treatments during dox pulse with no chase for regeneration or re-equilibration (2), interventions during pulse with an opportunity to visualize long-term effects (3). A short pulse of 2 days with no subsequent chase will provide insight into a near-basal state of TERT+ cells, as these cells will have little time to activate, proliferate, migrate, or differentiate (4). Please click here to view a larger version of this figure.
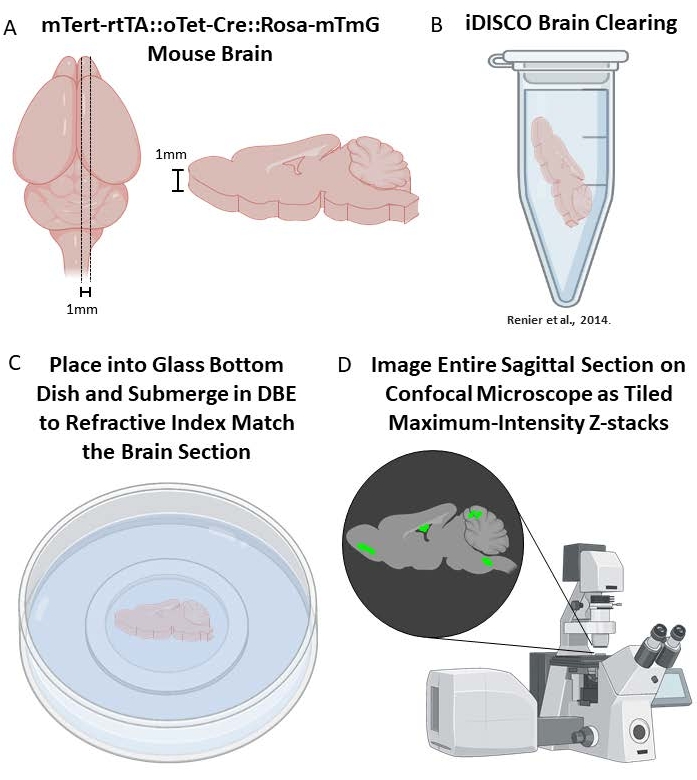
Figure 3. Brain clearing of mTert-rtTA::oTet-Cre::Rosa-mTmG mouse brains. (A) Sagittal sectioning of 1 mm brain sections after fixation. (B) Individual brain sections are placed in 5mL tubes for iDISCO brain clearing39. (C) Cleared brains are placed in glass bottom dishes and submerged in DBE for refractive index matching during image acquisition. (D) Capture the entire area of the brain as a series of Z-stacks that will be tiled together to form one comprehensive 3D image of the brain. This can then be Z-maximum intensity projected to create a 2D image from the 3D dataset. Please click here to view a larger version of this figure.
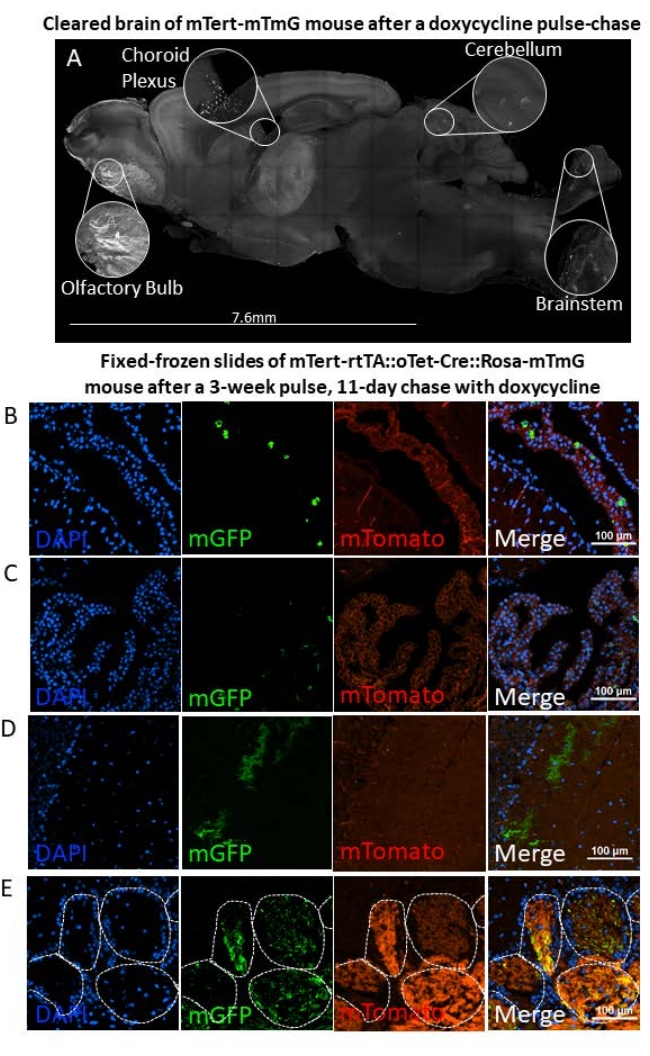
Figure 4. Lineage tracing of adult mTert-rtTA::oTet-Cre::Rosa-mTmG mouse brains after a 3 week pulse and 11 day chase of doxycycline. (A) Cleared brain section of adult mTert-rtTA::oTet-Cre::Rosa-mTmG mouse with specific areas of mGFP signal shown with insets (N = 3 male mice). (B–C) Representative image of mTert-rtTA::oTet-Cre::Rosa-mTmG choroid plexus identifying mGFP+ CPECs (B) and smaller, fainter mGFP+ cell types (C; N = 4 males, N = 6 females). (D) Representative image of mTert-rtTA::oTet-Cre::Rosa-mTmG cerebellar Bergmann glia (bright) and basket cells in the molecular layer of the cerebellum (dim; N = 4 males, N = 6 females). (E) Representative image of the olfactory glomerular layer of mTert-rtTA::oTet-Cre::Rosa-mTmG brains. Dotted lines indicate glomerular compartments (N = 4 males, N = 6 females). Scale bars are 100 µm. Please click here to view a larger version of this figure.
Table 1. Please click here to download this Table.
Supplementary Table 1. Please click here to download this Table.
Supplementary Table 2. Please click here to download this Table.
Discussion
A method for the creation, utilization, and analysis of a triple transgenic lineage tracing mouse line marking stem cells within the adult mouse brain in vivo is described. When combined with immunostaining or brain clearing, the identification and characterization of traced fluorescent cells across the brain can be accomplished. This technique offers the ability to understand the plastic/regenerative/remodeling potential of labeled stem cells as they activate, migrate, differentiate, or proliferate. While previous data obtained via label retention with thymidine-H3 and BrdU concluded that the V-SVZ, olfactory bulb, and subgranular zone of the dentate gyrus were the only neurogenic brain regions in adult mammals2,3,4,5,6,7,8,43, it is now understood that adult neurogenesis also occurs in the cortex44, hypothalamus45,46, and striatum47,48,49, as well as potentially other novel niches that have not been well-studied. Adult gliogenesis, the process by which glial precursor cells create newborn astrocytes and oligodendrocytes, occurs throughout the brain as well37, and glia and neurons are hypothesized to derive from the same multipotent stem cell. For these reasons, lineage tracing studies have been integral in understanding the role of various stem cell types that contribute to replenishing and regenerating neurons and glia in the adult mammalian brain (reviewed in50).
For studies in adult brain plasticity, the optimal age is 12 weeks of age, when the murine brain has completed development51. When planning the length of the doxycycline pulse and subsequent chase, an understanding of the process of interest is crucial, so that the pulse-chase timing is long enough to allow for the processes of interest to occur. For example, the creation of newborn neurons in the olfactory bulb from adult neural stem cells (ANSCs) in the V-SVZ is approximately 4 weeks, as determined by previous lineage tracing studies52. A pulse-chase study that will label the ANSCs in the V-SVZ must therefore be within that timeframe to allow for the ANSCs in the V-SVZ to divide, for these ANSCs to differentiate into intermediate cell types, and for these intermediate cell types to migrate to the olfactory bulb and differentiate into adult-born neurons. The length of the chase will determine the amount of time that labeled cells and their progeny must continue their migration and differentiation, although these processes will occur during the pulse as well. Taken together, it is important to understand the processes involved, as the timeline of each lineage tracing experiment must allow for the proper processes to occur.
While this manuscript details the Tet-On system, other lineage tracing transgenes exist that may be utilized. The CreERT2 transgene allows for the expression of a Cre recombinase that is only active in the presence of tamoxifen or 4-OHT, a tamoxifen derivative. When this Cre is regulated by a promoter of interest, 4-OHT or tamoxifen administration will only produce Cre recombination in cells that express that promoter. When paired with a reporter gene such as Rosa-LSL-GFP or Rosa-mTmG, Cre-lox recombination will indelibly mark the cells that express Cre during tamoxifen administration. One downside of this system is the use of tamoxifen, which has long-lasting adverse effects on neurogenesis in both prenatal and adult brains17. For this reason, lineage tracing studies with CreERT2 lines will need to consider the caveats involved.
Lineage tracing studies in the adult brain are often performed with markers of stem cells such as Nestin or markers of glial precursor cells including NG253,54. While resulting data indicates the turnover and differentiation of these cells into newborn mature cell types, the expression of these markers by various other cell types in the adult brain can be confounding. For example, Nestin, an intermediate filament protein involved in mitosis55, is also expressed by mature neurons56 and meningeal cell types57. Other stem cell markers include glial fibrillary acidic protein (GFAP)58,59, and glutamate aspartate transporter 1 (GLAST)60, which are expressed by astrocytes throughout the adult brain as well61. For this reason, lineage tracing studies with additional markers are needed. As we learn more about the broad impact of adult plasticity in the brain, the characterization and analysis of the cells that play a role in these processes and their progeny remain vitally important to our understanding of neurogenic and gliogenic pathways involved in neurodegenerative health and more.
Divulgations
The authors have nothing to disclose.
Acknowledgements
The authors wish to thank Dr. Diana Carlone (Boston Children's Hospital, Harvard Medical School) and Dr. Matthew Lynes (Joslin Diabetes Center; Maine Medical Center Research Institute) for guidance utilizing mTert-rtTA animals.
Materials
| Antibody diluent | Agilent | S080983-2 | |
| Antigen Retrieval Solution | Agilent | S2367 | |
| anti-GFP antibody | Invitrogen | A2311 | Use at 1:500-1:000 |
| Acetone | Fisher Scientific | A18-4 | |
| B6.Cg-Tg(tet0-cre)1Jaw/J | The Jackson Laboratory | JAX:006234 | |
| B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J | The Jackson Laboratory | JAX:007676 | |
| Blocking Solution (IHC) | Millipore Sigma | 20773 | |
| Blunt needle | BSTEAN | X0012SYHIV | |
| Coronal brain block | Braintree | BS-2000C | |
| Cover slip | Corning | 2850-22 | |
| Cryostat | Leica | CM1900 | |
| DAPI | Sigma-Aldrich | D564 | |
| Dibenzyl ether | Millipore Sigma | 33630 | |
| Dichloromethane | Sigma-Aldrich | 270997 | |
| Dimethyl Sulfoxide | Sigma-Aldrich | D8418 | |
| Donkey Serum | Sigma-Aldrich | D9663 | |
| Doxycycline Hyclate | Sigma-Aldrich | D9891 | |
| Glycine | Bio-Rad | 161-0718 | |
| Heparin Sodium Salt from Porcine Mucosa | Sigma-Aldrich | H3393 | |
| Histochoice Molecular Biology Tissue Fixative | Amresco | H120 | This product has since been discontinued, but can be replaced with Glyo-Fixx (Thermo Scientific, 6764265) |
| Hydrogen Peroxide Solution | Sigma-Aldrich | 516813 | |
| IHC Select TBS Rinse Buffer | Millipore Sigma | 20845 | |
| Ketamine | Westward | 0143-95095-10 | This product requires a DEA license for storage and use. |
| Methanol | Fisher Scientific | A452-4 | |
| Microscope Slides | Fisherbrand | 12-550-15 | |
| Milipore Block | Millipore | 20773 | |
| Mounting medium | Millipore Sigma | 5013 | |
| Optimal Cutting Temperature Solution | Sakura | 4583 | |
| Paraformaldehyde | Sigma-Aldrich | P6148 | |
| PBS Solution (10X) | Teknova | P0496 | |
| Sagittal brain block | Braintree | RBM-2000S | |
| Saline | Braun | S8004-5264 | |
| Sucrose | Sigma-Aldrich | S7903 | |
| Sudan (Typogen) Black | Millipore Sigma | 199664 | |
| Tetrahydrofuran | Sigma-Aldrich | 186562 | |
| Tissue cartridge | Simport | M512 | |
| Triton X-100 | Bio-Rad | 1610407 | |
| Tween-20 | Millipore Sigma | 655205 | |
| Xylazine | AnaSed | sc-362950Rx |
References
- Carlone, D. L., et al. Telomerase expression marks transitional growth-associated skeletal progenitor/stem cells. Stem Cells. 39 (3), 296-305 (2021).
- Altman, J., Das, G. D. Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. Journal of Comparative Neurology. 124 (3), 319-335 (1965).
- Cameron, H. A., Gould, E. Adult neurogenesis is regulated by adrenal steroids in the dentate gyrus. Neurosciences. 61 (2), 203-209 (1994).
- Kaplan, M. S., Hinds, J. W. Neurogenesis in the adult rat: electron microscopic analysis of light radioautographs. Science. 197 (4308), 1092-1094 (1977).
- Gould, E., et al. Neurogenesis in the dentate gyrus of the adult tree shrew is regulated by psychosocial stress and NMDA receptor activation. Journal of Neuroscience. 17 (7), 2492-2498 (1997).
- Eriksson, P. S., et al. Neurogenesis in the adult human hippocampus. Nature Medicine. 4 (11), 1313-1317 (1998).
- Gould, E., et al. Neurogenesis in adulthood: a possible role in learning. Trends in Cognitive Science. 3 (5), 186-192 (1999).
- Hastings, N. B., Gould, E. Rapid extension of axons into the CA3 region by adult-generated granule cells. Journal of Comparative Neurology. 413 (1), 146-154 (1999).
- Hu, V. W., et al. 3H-thymidine is a defective tool with which to measure rates of DNA synthesis. The FASEB Journal. 16 (11), 1456-1457 (2002).
- Cifuentes, M., et al. A comparative analysis of intraperitoneal versus intracerebroventricular administration of bromodeoxyuridine for the study of cell proliferation in the adult rat brain. Journal of Neuroscience Methods. 201 (2), 307-314 (2011).
- Taupin, P. BrdU immunohistochemistry for studying adult neurogenesis: paradigms, pitfalls, limitations, and validation. Brain Research Reviews. 53 (1), 198-214 (2007).
- Zeng, C., et al. Evaluation of 5-ethynyl-2′-deoxyuridine staining as a sensitive and reliable method for studying cell proliferation in the adult nervous system. Brain Research. 1319, 21-32 (2010).
- Guizzetti, M., Kavanagh, T. J., Costa, L. G. Measurements of astrocyte proliferation. Methods in Molecular Biology. 758, 349-359 (2011).
- Gomez-Nicola, D., et al. Regulation of microglial proliferation during chronic neurodegeneration. Journal of Neuroscience. 33 (6), 2481-2493 (2013).
- Bradl, M., Lassmann, H. Oligodendrocytes: biology and pathology. Acta Neuropathologica. 119 (1), 37-53 (2010).
- Das, A. T., Tenenbaum, L., Berkhout, B. Tet-On systems for doxycycline-inducible gene expression. Current Gene Therapy. 16 (3), 156-167 (2016).
- Lee, C. M., et al. Single-cell RNA-seq analysis revealed long-lasting adverse effects of tamoxifen on neurogenesis in prenatal and adult brains. Proceedings of the National Academy of Sciences. 117 (32), 19578-19589 (2020).
- Smith, B. M., et al. Oral and injected tamoxifen alter adult hippocampal neurogenesis in female and male mice. eNeuro. 9 (2), (2022).
- Greenberg, R. A., et al. Expression of mouse telomerase reverse transcriptase during development, differentiation and proliferation. Oncogene. 16 (13), 1723-1730 (1998).
- Cong, Y. S., Wright, W. E., Shay, J. W. Human telomerase and its regulation. Microbiology and Molecular Biology Reviews. 66 (3), 407-425 (2002).
- Breault, D. T., et al. Generation of mTert-GFP mice as a model to identify and study tissue progenitor cells. Proceedings of the National Academy of Sciences. 105 (30), 10420-10425 (2008).
- Montgomery, R. K., et al. Mouse telomerase reverse transcriptase (mTert) expression marks slowly cycling intestinal stem cells. Proceedings of the National Academy of Sciences. 108 (1), 179-184 (2011).
- Lin, S., et al. Distributed hepatocytes expressing telomerase repopulate the liver in homeostasis and injury. Nature. 556 (7700), 244-248 (2018).
- Lynes, M. D., et al. Telomerase reverse transcriptase expression marks a population of rare adipose tissue stem cells. Stem Cells. 40 (1), 102-111 (2022).
- Cousins, F. L., et al. Telomerase reverse transcriptase expression in mouse endometrium during reepithelialization and regeneration in a menses-like model. Stem Cells and Development. 28 (1), 1-12 (2019).
- Deane, J. A., et al. The mouse endometrium contains epithelial, endothelial and leucocyte populations expressing the stem cell marker telomerase reverse transcriptase. Molecular Human Reproduction. 22 (4), 272-284 (2016).
- Thompson, C. A. H., Wong, J. M. Y. Non-canonical functions of telomerase reverse transcriptase: emerging roles and biological relevance. Current Topics in Medicinal Chemistry. 20 (6), 498-507 (2020).
- Lynes, M. D., et al. Telomerase reverse transcriptase expression marks a population of rare adipose tissue stem cells. Stem Cells. , (2021).
- Redelsperger, I. M., et al. Stability of doxycycline in feed and water and minimal effective doses in tetracycline-inducible systems. Journal of the American Association for Laboratory Animal Science. 55 (4), 467-474 (2016).
- Jensen, G. J., et al. Telomerase reverse transcriptase (TERT)-expressing cells mark a novel stem cell population in the adult mouse brain. Révision par les pairs. , (2022).
- Muzumdar, M. D., et al. A global double-fluorescent Cre reporter mouse. Genesis. 45 (9), 593-605 (2007).
- Bocker, R., et al. Comparison of distribution of doxycycline in mice after oral and intravenous application measured by a high-performance liquid chromatographic method. Arzneimittelforschung. 31 (12), 2116-2117 (1981).
- Lucchetti, J., et al. Plasma and brain concentrations of doxycycline after single and repeated doses in wild-type and APP23 mice. Journal of Pharmacology and Experimental Therapeutics. 368 (1), 32-40 (2019).
- Sun, Y., Chen, X., Xiao, D. Tetracycline-inducible expression systems: new strategies and practices in the transgenic mouse modeling. Acta Biochimica et Biophysica Sinica. 39 (4), 235-246 (2007).
- Lee, P., et al. Conditional lineage ablation to model human diseases. Proceedings of the National Academy of Sciences. 95 (19), 11371-11376 (1998).
- Shi, S. R., et al. Evaluation of the value of frozen tissue section used as "gold standard" for immunohistochemistry. American Journal of Clinical Pathology. 129 (3), 358-366 (2008).
- Delgado, A. C., et al. Release of stem cells from quiescence reveals gliogenic domains in the adult mouse brain. Science. 372 (6547), 1205-1209 (2021).
- Bratthauer, G. L. Preparation of frozen sections for analysis. Immunocytochemical Methods and Protocols. , 67-73 (2010).
- Renier, N., et al. iDISCO: a simple, rapid method to immunolabel large tissue samples for volume imaging. Cell. 159 (4), 896-910 (2014).
- Cawthorne, C., et al. Comparison of doxycycline delivery methods for Tet-inducible gene expression in a subcutaneous xenograft model. Journal of Biomolecular Techniques. 18 (2), 120-123 (2007).
- Hristovska, I., et al. Ketamine/xylazine and barbiturates modulate microglial morphology and motility differently in a mouse model. PLoS One. 15 (8), 0236594 (2020).
- Pereira, G. C., et al. Anesthesia can alter the levels of corticosterone and the phosphorylation of signaling molecules. BMC Research Notes. 14 (1), 363 (2021).
- Markakis, E. A., Gage, F. H. Adult-generated neurons in the dentate gyrus send axonal projections to field CA3 and are surrounded by synaptic vesicles. Journal of Comparative Neurology. 406 (4), 449-460 (1999).
- Magavi, S. S., Leavitt, B. R., Macklis, J. D. Induction of neurogenesis in the neocortex of adult mice. Nature. 405 (6789), 951-955 (2000).
- Evans, J., et al. Characterization of mitotic neurons derived from adult rat hypothalamus and brain stem. Journal of Neurophysiology. 87 (2), 1076-1085 (2002).
- Kokoeva, M. V., Yin, H., Flier, J. S. Neurogenesis in the hypothalamus of adult mice: potential role in energy balance. Science. 310 (5748), 679-683 (2005).
- Parent, A., Cicchetti, F., Beach, T. G. Calretinin-immunoreactive neurons in the human striatum. Brain Research. 674 (2), 347-351 (1995).
- Rich, E. L., Shapiro, M. Rat prefrontal cortical neurons selectively code strategy switches. Journal of Neuroscience. 29 (22), 7208-7219 (2009).
- Suzuki, S. O., Goldman, J. E. Multiple cell populations in the early postnatal subventricular zone take distinct migratory pathways: a dynamic study of glial and neuronal progenitor migration. Journal of Neuroscience. 23 (10), 4240-4250 (2003).
- Figueres-Onate, M., Sanchez-Gonzalez, R. -., Lopez-Mascaraque, L. -. Deciphering neural heterogeneity through cell lineage tracing. Cellular and Molecular Life Sciences. 78 (5), 1971-1982 (2021).
- Hammelrath, L., et al. Morphological maturation of the mouse brain: An in vivo MRI and histology investigation. Neuroimage. 125, 144-152 (2016).
- Ming, G. L., Song, H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 70 (4), 687-702 (2011).
- Sanchez-Gonzalez, R., Bribian, A., Lopez-Mascaraque, L. Cell fate potential of NG2 progenitors. Scientific Reports. 10 (1), 9876 (2020).
- Suzuki, T., et al. Cells of NG2 lineage increase in glomeruli of mice following podocyte depletion. American Journal of Physiology – Renal Physiology. 315 (5), 1449-1464 (2018).
- Chou, Y. H., et al. Nestin promotes the phosphorylation-dependent disassembly of vimentin intermediate filaments during mitosis. Molecular Biology of the Cell. 14 (4), 1468-1478 (2003).
- Hendrickson, M. L., et al. Expression of nestin by neural cells in the adult rat and human brain. PLoS One. 6 (4), 18535 (2011).
- Bifari, F., et al. Meninges harbor cells expressing neural precursor markers during development and adulthood. Frontiers in Cellular Neuroscience. 9, 383 (2015).
- Doetsch, F., et al. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 97 (6), 703-716 (1999).
- Seri, B., et al. Astrocytes give rise to new neurons in the adult mammalian hippocampus. Journal of Neuroscience. 21 (18), 7153-7160 (2001).
- DeCarolis, N. A., et al. In vivo contribution of nestin- and GLAST-lineage cells to adult hippocampal neurogenesis. Hippocampus. 23 (8), 708-719 (2013).
- Ihrie, R. A., Alvarez-Buylla, A. Cells in the astroglial lineage are neural stem cells. Cell and Tissue Research. 331 (1), 179-191 (2008).
