The Specification of Telencephalic Glutamatergic Neurons from Human Pluripotent Stem Cells
Summary
This procedure yields telencephalic neurons by going through checkpoints which are similar to those observed during human development. The cells are allowed to spontaneously differentiate, are exposed to factors which push them towards the neural lineage, are isolated, and are plated onto coverslips to allow for terminal differentiation and maturation.
Abstract
Here, a stepwise procedure for efficiently generating telencephalic glutamatergic neurons from human pluripotent stem cells (PSCs) has been described. The differentiation process is initiated by breaking the human PSCs into clumps which round up to form aggregates when the cells are placed in a suspension culture. The aggregates are then grown in hESC medium from days 1-4 to allow for spontaneous differentiation. During this time, the cells have the capacity to become any of the three germ layers. From days 5-8, the cells are placed in a neural induction medium to push them into the neural lineage. Around day 8, the cells are allowed to attach onto 6 well plates and differentiate during which time the neuroepithelial cells form. These neuroepithelial cells can be isolated at day 17. The cells can then be kept as neurospheres until they are ready to be plated onto coverslips. Using a basic medium without any caudalizing factors, neuroepithelial cells are specified into telencephalic precursors, which can then be further differentiated into dorsal telencephalic progenitors and glutamatergic neurons efficiently. Overall, our system provides a tool to generate human glutamatergic neurons for researchers to study the development of these neurons and the diseases which affect them.
Introduction
Human pluripotent stem cells (PSCs), including both human embryonic stem cells (hESCs) and induced pluripotent stem cells (iPSCs), have the capacity to generate every cell type in the body, including neurons1-3. Directed differentiation of various neuronal subtypes from human PSCs holds the key for the application of these cells in regenerative medicine. The generation of functional neuronal subtypes during development is a complex process involving the induction of neural lineage, the specification of regional progenitors along the rostro-caudal axis, and the differentiation of post-mitotic neuron types from the regional progenitors4,5. Beginning in 2001, several systems have been established to generate neural lineage from hESCs, which have provided a platform for the subsequent generation of neuronal subtypes6,7. Based upon developmental principles, several neuron types such as spinal motor neurons8-12, midbrain dopaminergic neurons13-15, and neural retinal cells16,17 have been efficiently specified from human PSCs. This was done by applying critical morphogens which are important for the specification of these neuron types during in vivo development. Other protocols have also been developed to promote the differentiation of hESCs into neurons using either additional factors18-20 such as small molecules or by co-culturing with other cell types to help promote differentiation21.
The human neocortex is highly developed and contains many cell types, including glutamatergic neurons which play an important role in learning, memory, and cognitive function22,23. The first step in generating glutamatergic neurons in culture is to specify telencephalic progenitor cells. Yoshiki Sasai’s group first reported the directed differentiation of telencephalic precursors from mouse ESCs (mESCs) using a serum-free suspension culture in the presence of DKK1 (which inhibits Wnt signaling) as well as LeftyA (which inhibits nodal signaling)24. Subsequently, several groups including ours have also reported the specification of telencephalic precursors from human PSCs in serum free medium 25-27. The generation of telencephalic precursors from human PSCs does not require the use of exogenous morphogens and the efficiency in generating these precursors is much higher than that from mESCs 26,27. Here, a chemically defined system for neural induction which was well established by Zhang’s group7 has been described. Without the addition of exogenous caudalizing factors, this protocol efficiently generates telencephalic precursors from human PSCs27. These progenitors can then be differentiated into dorsal or ventral progenitors by regulating the signaling of Wnt and sonic hedgehog (SHH).The dorsal progenitors can further differentiate into glutamatergic neurons efficiently27. In addition, this protocol also works well for the generation of glutamatergic neurons from human iPSCs28, which allows for the generation of patient-specific neurons that can be utilized to explore the mechanism of action as well as potential therapies for a large array of diseases. Moreover, our system also provides a platform to explore the development and specification of diverse neuronal types in the telencephalon.
Protocol
1. Generation of Human Pluripotent Stem Cell Aggregates (D1-D4)
- Human pluripotent stem cells are cultured on mouse embryonic fibroblast (MEF) feeders in the presence of hESC medium supplemented with basic fibroblast growth factor (bFGF, 4 ng/ml). After 5-7 days in culture, when the colonies are big but still undifferentiated, they are ready for the next step.
- The enzyme solution should first be prepared. In a 50 ml tube, dissolve the dispase (or collagenase) at a 1 mg/ml concentration into DMEM/F12 medium. Since these solutions mix best when warmed, place the tube containing the medium and enzyme into a 37 °C water bath for 10-15 min and then sterilize it using a Steriflip filter.
- Aspirate off the medium from the cells, rinse them with DMEM/F12, and aspirate this off as well. Add 1 ml of dispase to each well and place in the incubator for 3-5 min for hESCs (up to 10 min for iPSCs). Look at the cells under the microscope; when the edges curl up slightly/look a bit darker, the cells are ready for the next step. It is best to check the cells after 3 min and put them back in if necessary. When using this protocol for the first time, it is best to start with only one plate of cells at a time as it is important to remove the cells from the dispase as quickly as possible.
- Aspirate off the dispase from the cells. Gently (the cells are very vulnerable at this stage and will come off of the plate relatively easily) add 1.5 ml of DMEM/F12 medium to each well in order to rinse off the dispase. Aspirate off the DMEM/F12 and add 1.5 ml of hESC medium to each well.
- To detach and break up the cells, place the tip of a 10 ml glass pipette towards the bottom right hand corner of a well (touching the cells) and move the pipette up and down (vertically) – the upward stoke should be in close proximity to the proceeding downward stroke. Once the other side of the well is reached, repeat in the horizontal direction.
- Once all of the cells are in suspension, place them in 15 ml tubes and centrifuge them at 200 x g for 2 min. There should be a pellet of cells at the bottom of the tube. Aspirate off the medium from above the pellet.
- The size of the cell culture flask that should be utilized to keep the aggregates in depends upon the original quantity of cells. For 6 wells (1 plate) of hESCs, one non-tissue culture treated T75 flask with 50 ml of hESC medium should be utilized. Transfer the cells from step 1.6 to the flask and incubate at 37 °C.
- In order to get rid of the cell debris, the cell medium and flask should be replaced approximately 12 hr later. To do this, place all of the medium/cells into a 50 ml tube, spin the cells down (200 x g for 2 min), and aspirate off the old medium. Next, add 50 ml of fresh hESC medium to the cells, and place into a new T75 flask. The cells can then be fed daily following this same procedure (until day 5).
2. Induction of Neuroepithelial Cells (D5-D17)
- Transfer the cells/medium to a 50 ml tube, and centrifuge the cells for 2 min at 200 x g. Next, aspirate off the supernatant, and transfer the cells to a new (non-tissue culture treated) T75 flask with 50 ml of neural induction medium (NIM).
- Once the cells are in NIM, half of the medium should be replaced every other day. The aggregates should become large enough to where they will sink to the bottom of a 50 ml conical tube on their own after a few minutes (no centrifugation required). After 2-3 days in NIM (D7-D8 total), the cells should be ready to plate. Laminin or fetal bovine serum (FBS) can be utilized to help the cells attach to the plates.
- The number of tissue culture treated 6 well plates that are needed depends upon the density of the cells as each aggregate should be able to grow on the plate for several more days without touching any of the others. On average, the cells from one T75 flask will yield about 4-6 6 well plates worth of cells.
-
- If using laminin, dilute it in NIM to 10-20 μg/ml to coat the 6-well plate (~0.5 ml/well). After coating for 2-3 hr, plate cell aggregates onto 6-well plates (~20-30 aggregates per well). After the aggregates have attached, add 1.5 ml of NIM.
- If using FBS, prepare a mixture of 90% NIM and 10% FBS (1.5 ml/well). Add 1.5 ml of medium with the cells to each well, gently shake the plate back and forth to spread out the aggregates, and allow the cells to attach O/N in an incubator. Be sure to change the medium (2 ml/well of NIM) the following morning to remove the FBS.
- After a day or two of being plated, each aggregate will spread out to form a monolayer. Within a couple of days, the centers of most of these cells will morphologically appear similar to columnar cells (elongated). During this period, change the NIM every other day. Some PSC lines (especially iPSC), may form columnar cells less efficiently. If this happens, the addition of BMP and TGF inhibitors (Dorsomorphin29, SB43154219 at 2 μM) during stage 2 can be helpful.
- Around days 14-17, the columnar cells will appear more compact. Sometimes these cells form ridges or rings with visible lumen inside. Besides the columnar morphology, cells at this stage express neuroepithelial markers, such as PAX6 and SOX112,26.
- Before the isolation can begin, one must first examine the cells to determine if there are any non-neuroepithelial cells present. An objective marker can be utilized to indicate the non-neuroepithelial colonies. These can then be eliminated by scraping them off with a pipette tip.
3. Generation of Telencephalic Progenitors (D17-D24)
- The centers of the colonies contain the neuroepithelial cells, and should be isolated from the rest of the colony. This can be done using a micropipette and a sterile 1 ml filtered pipette tip. When a small amount of pressure is applied to the center portion of the colony, it generally lifts off quite readily. Be cautious to not let the outer portion of the colony detach as well. If the centers are being stubborn and will not come off on their own, they can be removed by excising them using a needle under a microscope in a sterile hood. Transfer the center portions of the colonies to a 15 ml tube and spin at 200 x g for 2 min.
- The medium should be aspirated off of the cells. Two plates worth of cells can then be transferred to a non-tissue culture treated T25 flask containing 10 ml of NIM with the addition of B27 (without vitamin A, 1:50). Use 5-10 ml of medium per one plate of cells.
- When the cells are viewed the following day, they should have a sphere like appearance. It is necessary to transfer these cells into a new flask (in the presence of NIM and B27, 1:50) as it is often the peripheral cells which sneak in and attach to the bottom of the flask.
- The neurospheres should then be cultured in suspension for one week using NIM supplemented with B27 (1:50). In order to promote cell endurance and proliferation, cyclic AMP (cAMP, 1 μM) as well as insulin growth factor (IGF1, 10 ng/ml) can be added into the medium during suspension culture. At this point, the neurospheres contain telencephalic progenitors (FOXG1+) and are ready to be plated onto coverslips for terminal differentiation.
4. Further Differentiation into the Telencephalic Neurons (D25+)
-
- Prepare the poly-ornithine/laminin coated coverslips (in 24-well plates) for plating the cells. Coverslips are first cleaned, and are then coated with ploy-ornithine (at 0.1 mg/ml, 37 °C, O/N, and stored in the freezer).
Poly-ornithine coating:- Sterilize the coverslips: Place the coverslips in a beaker containing nitric acid and gently shake in the fume hood for one hour. Pour out the nitric acid, rinse several times with DI water, and leave the coverslips under running DI water for at least 15 min. Place the coverslips in 95% ethanol and either shake for 30 min (if using immediately) or store in ethanol at RT.
- In a sterile hood, pour out the contents of the 50 ml tube containing sterilized coverslips into the lid of a 6-well plate. Using sterilized forceps, pick up one coverslip at a time and set upright in a single well of a 24-well plate. Repeat so that each well has a coverslip.
- After they dry completely, tap the plates until coverslips have fallen flat in each well. Next add 100 μl poly-ornithine (0.1 mg/ml in sterile dH2O) to each coverslip and incubate the plates O/N at 37 °C.
- The next day, remove plates from the incubator and allow them to cool to RT. Aspirate poly-ornithine off of each coverslip (from the edge of the coverslip) making sure not to touch or scratch the coverslips in the process. Allow the coverslips to dry for approximately 30 min before washing.
- Add 1 ml sterile dH2O to each well, let sit for 10 min, and aspirate off the water from each well. Repeat this wash step two more times. After the plates dry completely, (leave covers off in the hood) place the lids on, cover with aluminum foil, and store at -20 °C.
-
- To coat the coverslips with laminin, add 50 μl of a mixture of neural differentiation medium and laminin (final concentration of 20 μg/ml) to each poly-ornithine treated coverslip being careful to only have the liquid touching the coverslip itself (and not spilling off). Incubate them at 37 °C for 1-2 hr before preparing the cells for attachment.
- Prepare the poly-ornithine/laminin coated coverslips (in 24-well plates) for plating the cells. Coverslips are first cleaned, and are then coated with ploy-ornithine (at 0.1 mg/ml, 37 °C, O/N, and stored in the freezer).
- Collect and centrifuge the neurospheres at 200 x g for 2 min in a 15 ml tube. Aspirate off the supernatant.
-
- Rinse the cells with DMEM-F12 and centrifuge the cells again. If the neurospheres are too large to attach properly, they can be dissociated with Accutase at this step.
- If using Accutase: After washing the cells with DMEM-F12, the neurospheres are dissociated to small clusters by adding Accutase (~2 ml) and incubating the cells at 37 °C for 3 min. Next, add the same amount of trypsin inhibitor (~2 ml) and centrifuge the cells at 200 x g for 3 min. The cells should then be re-suspended in NDM and broken using a P200 pipette. The small clusters can then be plated onto pre-coated coverslips.
- Aspirate off the majority of the medium and place the cells onto a 6 cm Petri dish in a few drops of the medium so that selecting them becomes easier. Add about 4-5 neurospheres to each pre-coated coverslip. There is no need to aspirate off the laminin first.
- Let the neurospheres attach for at least 2-4 hr. Once they have attached well, more medium (0.5 ml/coverslip) should be added. It should consist of neural differentiation medium supplemented with B27 (1:50), cAMP (1 μM), and neurotrophic factors (BDNF, GDNF, and IGF, all at 10 ng/ml). At this point half of the medium can be changed every other day and these cells can be maintained for several months.
Representative Results
Here, a protocol to differentiate human PSCs into telencephalic glutamatergic neurons through several critical steps: the formation of PSC aggregates, the induction of neuroepithelial cells, the generation of telencephalic progenitors, and the terminal differentiation of these progenitors into telencephalic neurons (Figure 1) has been described. This system is robust and efficient in the generation of telencephalic progenitors and glutamatergic neurons. As an example (Figure 2), without the addition of caudalizing factors, the hESCs were differentiated into the neural lineage27. At 24 days post differentiation, the majority of neural precursors were positive for FOXG1 (a transcription factor expressed by telencephalon) but negative for HOXB4 (a marker for hindbrain and spinal cord cells) suggesting the telencephalic progenitors were successfully generated (Figure 2D). These telencephalic progenitors possess a dorsal phenotype, as indicated by the expression of PAX6 (a marker expressed by dorsal progenitors) (Figure 2E), but not NKX2.1 (a marker for ventral progenitors) (Figure 2F). Following further differentiation, these telencephalic dorsal progenitors differentiated into glutamatergic neurons, which were positive for the glutamatergic marker TBR1 (around 80%)27. Neurons which stained positive for TBR1 were also positive for neuronal markers βIII-tubulin (Figure 2G) or microtubule-associated protein 2 (MAP2, Figure 2H). These cells also expressed vesicular glutamate transporter 1 (vGLUT1), a marker for mature glutamatergic neurons, suggesting the efficient generation of telencephalic glutamatergic neurons in the culture (Figure 2I).
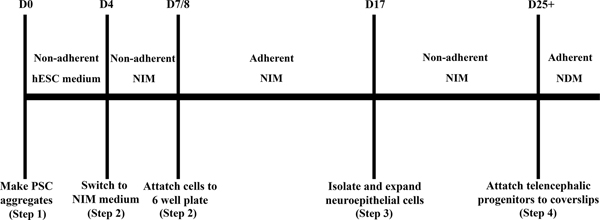
Figure 1. A schematic timeline for the neuronal differentiation process.
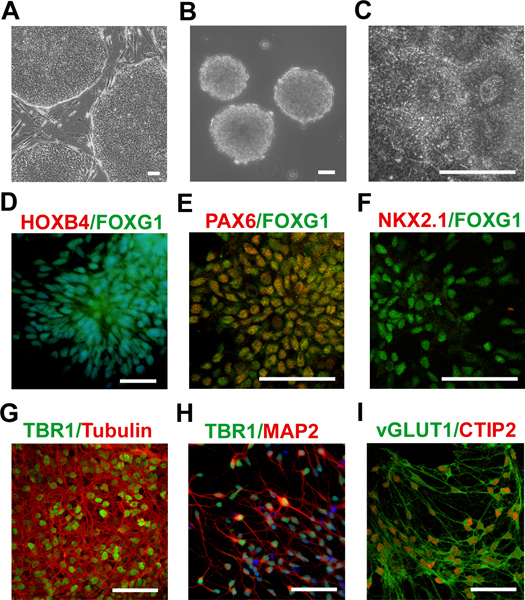
Figure 2. Images highlighting cells during several critical stages during neuronal differentiation. (A) Human ESCs were cultured for 4 days. Phase images showing the formation of ESC aggregates (B) and neuroepithelial cells (C). At 24 days after differentiation from hESCs, the majority of NE cells were FOXG1+ but HOXB4– (D). The telencephalic progenitors (FOXG1+) were PAX6+ (E) but NKX2.1– (F) after 1 month of differentiation. After two additional weeks (6 weeks total) of differentiating the cells on the coverslips, most of them stained positive for the glutamatergic marker TBR1 (G,H) as well as the neuronal markers βIII-tubulin (G) and MAP2 (H). After eight weeks of differentiation, the cells were positive for the mature glutamatergic marker, vGLUT1 (I). Scale bars: 100 μm (A), 50 μm (B-I). (D-I have been adapted from our previous publication27 with permission).
Discussion
There are several critical steps during the neural differentiation process. It is important to ensure that the human PSCs are pluripotent because otherwise the cells may already be biased towards becoming a non-neuronal lineage. This can be confirmed by staining the human PSCs with antibodies against pluripotency markers such as Oct4, Sox2, Nanog, and Tra-1-60 1-3. If the human PSCs do not attach very well after passaging them, ROCK inhibitor (Y27632) can be added to help. For those having difficulty with keeping their cells pluripotent, some potential issues are the quality/density of the MEF cells, as well as the lot of KOSR being used as there can be variation from batch to batch. Although MEF feeder cells have been used to maintain PSCs in the above protocol, this method also works for PSCs that are cultured using feeder-free systems.
When the cells are being broken up to form the PSC aggregates, it is important to limit their dispase exposure to only a few minutes (the edges will round up). The length of time that it takes before the edges of the iPSCs (up to 10 min) is usually longer than that of the ESCs (3-5 min). It is optimal to add the dispase to only one plate of human PSCs at a time to ensure that the cells are not in the enzyme for too long as this can harm or even kill the cells. After the cells are in the PSC aggregate stage, it is important to keep the cells at a low density. The density is also quite important when the PSC aggregates are plated. These cells will expand on the plate over the next few days and precautions should be taken to ensure that they will not touch after they grow bigger. During the attachment step (2.2), it is imperative to ensure that the cells do not have a prolonged exposure to the FBS as this could affect the gene expression. Before isolating the neuroepithelial cells, it is also important to scratch off the non-neuronal clusters in order to yield a more pure population of cells. If one wanted to characterize whether or not their cells are going into the neuronal lineage, various factors can be looked at such as Pax6 or Sox1. Pax6 turns on around day 10 during neural differentiation, and Sox1 turns on by 2 weeks after differentiation12,26,30.
This protocol is at the forefront of neural differentiation as it recapitulates many steps that take place during the development of the human nervous system. Without the use of caudalizing factors (retinoic acid, basic FGF), neuroepithelial cells efficiently differentiate into telencephalic progenitors, which coincides with neuroectoderm cells first acquiring a rostral phenotype during in vivo development31. These telencephalic progenitors possess a dorsal phenotype due to endogenous Wnt signaling which dorsalizes the cells27. This system generates dorsal progenitors which can then be further differentiated into glutamatergic cells. While this cell type is very important, it is by no means the only cell type that this system is capable of forming. For example, the addition of SHH has been shown to ventralize the cells, allowing them to differentiate into GABAergic cells27,32. It was even demonstrated that these hESC derived GABAergic neurons can be transplanted into mice and that they are able to correct locomotor defects due to brain lesions32. Because the protocol that is demonstrated in this article goes through the stepwise checkpoints, it offers a tool to produce a wide array of cells within the human nervous system which is merely limited by our understanding of development and our imaginations.
The ability to form human neurons from PSCs opens a large number of doors from both a basic science as well as a clinical perspective. Postmortem tissue sources are limited and the quality can vary, whereas human PSC differentiation allows researchers to yield an unlimited and consistent supply of cells to work with. With the advent of induced pluripotent stem cell (iPSC) technology1,3,33,34, it is now feasible to get hESC-like cells from patient fibroblasts including those with various diseases35-38. As shown by our group28 as well as many others, the successful generation of neurons from iPSC has been and will continue to be a unique and useful tool for those who have long sought after human models for disease and development. In addition, several groups have shown that PSC-derived neurons can model certain aspects of the disease process36,37,39-42 and thus can be utilized to screen therapeutic compounds43. This is particularly encouraging because without a good model system to test candidate drugs for the central nervous system with, only about 8% of them have been shown to be clinically effective44. However, this method and others like it could help to dramatically improve this number.
Divulgazioni
The authors have nothing to disclose.
Acknowledgements
The authors would like to thank Dr. Y. Sasai for generously providing the FOXG1 antibody. This work was supported by Connecticut Stem Cell Research Grants (08-SCB-UCHC-022 and 11-SCB24) and Spastic Paraplegia Foundation.
Materials
| Reagent | Supplier | Catalog # |
| Dulbecco’s modified eagle medium with F12 nutrient mixture (DMEM/F12) | Gibco | 11330-032 |
| Knockout Serum Replacer | Gibco | 10828-028 |
| L-glutamine (200 mM) | Gibco | 25030 |
| Non Essential Amino Acids | Gibco | 1140-050 |
| 2-Mercaptoethanol (14.3 M) | Sigma | M-7522 |
| Neurobasal medium | Gibco | 21103-049 |
| N2 | Gibco | 17502-048 |
| B27 | Gibco | 12587-010 |
| Heparin | Sigma | H3149 |
| Poly-L-ornithine hydrobromide (polyornithine) | Sigma | 116K5103 |
| Laminin (human) | Sigma | L-6274 |
| Laminin (mouse) | Invitrogen | 23017-015 |
| FBS | Gemini | 100-106 |
| Bovine serum albumin (BSA) | Sigma | A-7906 |
| Dispase | Gibco | 17105-041 |
| Collagenase | Invitrogen | 17104-019 |
| Accutase | Innovative Cell Technologies | AT104 |
| ROCK Inhibitor | Stemgent | 04-0012 |
| SB431542 | Stemgent | 04-0010 |
| Dorsomorphin | Stemgent | 04-0024 |
| Fibroblast growth factor 2 (FGF2, bFGF) | Invitrogen | 13256-029 |
| Trypsin inhibitor | Gibco | 17075 |
| 0.1% gelatin | Millipore | ES-006-B |
| Foxg1 antibody | Dr. Y. Sasai | |
| Hoxb4 antibody (1:50) | Developmental Studies Hybridoma Bank | I12 |
| Pax6 antibody (1:5000) | Developmental Studies Hybridoma Bank | PAX6 |
| Nkx2.1 antibody (1:200) | Chemicon | MAB5460 |
| Tbr1 antibody (1:2000) | Chemicon | AB9616 |
| vGLUT1 antibody (1:100) | Synaptic Systems | 135302 |
| Brain derived neurotrophic factor (BDNF) | PrepoTech Inc. | 450-02 |
| Glial derived neurotrophic factor (GDNF) | PrepoTech Inc. | 450-10 |
| Insulin growth factor 1 (IGF1) | PrepoTech Inc. | 100-11 |
| Cyclic AMP (cAMP) | Sigma | D-0260 |
| Sonic hedgehog (SHH) | R&D | 1845-SH |
| 50 ml tubes | Becton Dickinson (BD) | 352098 |
| 15 ml tubes | BD | 352097 |
| 6 well plates | BD | 353046 |
| 24 well plates | BD | 353047 |
| T25 flasks (untreated) | BD | 353009 |
| T75 flasks (untreated) | BD | 353133 |
| Coverslips | Chemiglass Life Sciences | 1760-012 |
| 6 cm Petri dishes | BD | 353004 |
| 9” glass pipetes | Fisher | 13-678-20D |
| Steriflip filters (0.22 μM) | Millipore | SCGP00525 |
| Stericup filters 1,000 ml (0.22 μM) | Millipore | SCGPU10RE |
| Phase contrast microscope (Observer A1) | Zeiss | R2625 |
| Carbon dioxide incubator (Hera Cell 150) | Thermo Electron Corporation | |
| Biosafety hood (Sterilgard III Advance) | The Baker Company | |
| Centrifuge (5702 R) | Eppendorf |
Riferimenti
- Takahashi, K., Tanabe, K., et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 131, 861-872 (2007).
- Thomson, J. A., Itskovitz-Eldor, J., et al. Embryonic stem cell lines derived from human blastocysts. Science. 282, 1145-1147 (1998).
- Yu, J., Vodyanik, M. A., et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 318, 1917-1920 (2007).
- Jessell, T. M. Neuronal specification in the spinal cord: inductive signals and transcriptional codes. Nat. Rev. Genet. 1, 20-29 (1038).
- Wilson, S. I., Edlund, T. Neural induction: toward a unifying mechanism. Nat. Neurosci. 4, 1161-1168 (2001).
- Reubinoff, B. E., Itsykson, P., et al. Neural progenitors from human embryonic stem cells. Nat. Biotechnol. 19, 1134-1140 (2001).
- Zhang, S. C., Wernig, M., Duncan, I. D., Brustle, O., Thomson, J. A. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat. Biotechnol. 19, 1129-1133 (2001).
- Hu, B. Y., Weick, J. P., et al. Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc. Natl. Acad. Sci. U.S.A. 107, 4335-4340 (2010).
- Singh Roy, N., Nakano, T., et al. Enhancer-specified GFP-based FACS purification of human spinal motor neurons from embryonic stem cells. Exp. Neurol. 196, 224-234 (2005).
- Lee, H., Shamy, G. A., et al. Directed differentiation and transplantation of human embryonic stem cell-derived motoneurons. Stem Cells. 25, 1931-1939 (2007).
- Boulting, G. L., Kiskinis, E., et al. A functionally characterized test set of human induced pluripotent stem cells. Nat. Biotechnol. 29, 279-286 (2011).
- Li, X. J., Du, Z. W., et al. Specification of motoneurons from human embryonic stem cells. Nat. Biotechnol. 23, 215-221 (2005).
- Perrier, A. L., Tabar, V., et al. Derivation of midbrain dopamine neurons from human embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 101, 12543-12548 (2004).
- Roy, N. S., Cleren, C., et al. Functional engraftment of human ES cell-derived dopaminergic neurons enriched by coculture with telomerase-immortalized midbrain astrocytes. Nat Med. 12, 1259-1268 (2006).
- Yan, Y., Yang, D., et al. Directed differentiation of dopaminergic neuronal subtypes from human embryonic stem cells. Stem Cells. 23, 781-790 (2005).
- Meyer, J. S., Shearer, R. L., et al. Modeling early retinal development with human embryonic and induced pluripotent stem cells. Proc. Natl. Acad. Sci. U.S.A. 106, 16698-16703 (2009).
- Osakada, F., Ikeda, H., et al. Toward the generation of rod and cone photoreceptors from mouse, monkey and human embryonic stem cells. Nat Biotechnol. 26, 215-224 (2008).
- Carpenter, M. K., Inokuma, M. S., et al. Enrichment of neurons and neural precursors from human embryonic stem cells. Exp. Neurol. 172, 383-397 (2001).
- Chambers, S. M., Fasano, C. A., et al. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 27, 275-280 (2009).
- Chambers, S. M., Qi, Y., et al. Combined small-molecule inhibition accelerates developmental timing and converts human pluripotent stem cells into nociceptors. Nat. Biotechnol. 30, 715-720 (2012).
- Kawasaki, H., Mizuseki, K., et al. Induction of midbrain dopaminergic neurons from ES cells by stromal cell-derived inducing activity. Neuron. 28, 31-40 (2000).
- Rubenstein, J. L., Beachy, P. A. Patterning of the embryonic forebrain. Curr. Opin. Neurobiol. 8, 18-26 (1998).
- Hevner, R. F., Hodge, R. D., Daza, R. A., Englund, C. Transcription factors in glutamatergic neurogenesis: conserved programs in neocortex, cerebellum, and adult hippocampus. Neurosci. Res. 55, 223-233 (2006).
- Watanabe, K., Kamiya, D., et al. Directed differentiation of telencephalic precursors from embryonic stem cells. Nat. Neurosci. 8, 288-296 (2005).
- Watanabe, K., Ueno, M., et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat. Biotechnol. 25, 681-686 (2007).
- Pankratz, M. T., Li, X. J., et al. Directed neural differentiation of human embryonic stem cells via an obligated primitive anterior stage. Stem Cells. 25, 1511-1520 (2007).
- Li, X. J., Zhang, X., et al. Coordination of sonic hedgehog and Wnt signaling determines ventral and dorsal telencephalic neuron types from human embryonic stem cells. Development. 136, 4055-4063 (2009).
- Zeng, H., Guo, M., et al. Specification of region-specific neurons including forebrain glutamatergic neurons from human induced pluripotent stem cells. PLoS One. 5, e11853 (2010).
- Kim, D. S., Lee, J. S., et al. Robust enhancement of neural differentiation from human ES and iPS cells regardless of their innate difference in differentiation propensity. Stem Cell Rev. 6, 270-281 (2010).
- Zhang, X., Huang, C. T., et al. Pax6 is a human neuroectoderm cell fate determinant. Cell Stem Cell. 7, 90-100 (2010).
- Stern, C. D. Initial patterning of the central nervous system: how many organizers. Nat. Rev. Neurosci. 2, 92-98 (2001).
- Ma, L., Hu, B., et al. Human embryonic stem cell-derived GABA neurons correct locomotion deficits in quinolinic acid-lesioned mice. Cell Stem Cell. 10, 455-464 (2012).
- Li, W., Wei, W., et al. Generation of rat and human induced pluripotent stem cells by combining genetic reprogramming and chemical inhibitors. Cell Stem Cell. 4, 16-19 (2009).
- Lowry, W. E., Richter, L., et al. Generation of human induced pluripotent stem cells from dermal fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 105, 2883-2888 (2008).
- Dimos, J. T., Rodolfa, K. T., et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 321, 1218-1221 (2008).
- Ebert, A. D., Yu, J., et al. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature. 457, 277-280 (2009).
- Lee, G., Papapetrou, E. P., et al. Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature. 461, 402-406 (2009).
- Park, I. H., Arora, N., et al. Disease-specific induced pluripotent stem cells. Cell. 134, 877-886 (2008).
- Chamberlain, S. J., Chen, P. F., et al. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes. Proc. Natl. Acad. Sci. U.S.A. 107, 17668-17673 (2010).
- Kiskinis, E., Eggan, K. Progress toward the clinical application of patient-specific pluripotent stem cells. J. Clin. Invest. 120, 51-59 (2010).
- Koch, P., Tamboli, I. Y., et al. Presenilin-1 L166P mutant human pluripotent stem cell-derived neurons exhibit partial loss of gamma-secretase activity in endogenous amyloid-beta generation. Am. J. Pathol. 180, 2404-2416 (2012).
- Walsh, R. M., Hochedlinger, K. Modeling Rett syndrome with stem cells. Cell. 143, 499-500 (2010).
- Egawa, N., Kitaoka, S., et al. Drug Screening for ALS Using Patient-Specific Induced Pluripotent Stem Cells. Sci. Transl. Med. 4, (2012).
- Kola, I., Landis, J. Can the pharmaceutical industry reduce attrition rates. Nature Reviews Drug Discovery. 3, 711-716 (2004).
