Application of an In vitro DNA Protection Assay to Visualize Stress Mediation Properties of the Dps Protein
Summary
The DNA-binding protein from starved cells (Dps) plays a crucial role in combating bacterial stress. This article discusses the purification of E. coli Dps and the protocol for an in vitro assay demonstrating Dps-mediated protection of DNA from degradation by reactive oxygen species.
Abstract
Oxidative stress is an unavoidable byproduct of aerobic life. Molecular oxygen is essential for terrestrial metabolism, but it also takes part in many damaging reactions within living organisms. The combination of aerobic metabolism and iron, which is another vital compound for life, is enough to produce radicals through Fenton chemistry and degrade cellular components. DNA degradation is arguably the most damaging process involving intracellular radicals, as DNA repair is far from trivial. The assay presented in this article offers a quantitative technique to measure and visualize the effect of molecules and enzymes on radical-mediated DNA damage.
The DNA protection assay is a simple, quick, and robust tool for the in vitro characterization of the protective properties of proteins or chemicals. It involves exposing DNA to a damaging oxidative reaction and adding varying concentrations of the compound of interest. The reduction or increase of DNA damage as a function of compound concentration is then visualized using gel electrophoresis. In this article we demonstrate the technique of the DNA protection assay by measuring the protective properties of the DNA-binding protein from starved cells (Dps). Dps is a mini-ferritin that is utilized by more than 300 bacterial species to powerfully combat environmental stressors. Here we present the Dps purification protocol and the optimized assay conditions for evaluating DNA protection by Dps.
Introduction
Aerobic organisms must constantly contend with reactive oxygen species that can damage their DNA as well as other crucial biological macromolecules. One potent tool to counteract the toxic effects of oxidative damage is the DNA-binding protein from starved cells (Dps). Since its discovery in 1992 from starved E. coli culture 1, Dps has been identified in more than 300 species of bacteria and archaebacteria 2. Massive upregulation of Dps during stationary phase makes it the most highly expressed nucleoid-associated protein of E. coli under starvation conditions 3, 4. Additionally, Dps has been shown to preserve both bacterial viability and DNA integrity during many various stresses, including starvation, high iron concentration, UV light exposure, heat shock, and oxidative stress 5, 6.
Structurally, Dps self-associates into a stable homo-oligomeric complex of 12 monomers, which assemble into a spherical hollow shell. The ~4.5 nm-wide internal cavity is accessible to the exterior solvent via pores that allow the passage of small molecules 7, and it can sequester mineralized metals such as iron 8. The protective effect of Dps derives from its several biochemical activities, which include non-specific DNA binding 1, ferroxidase activity, and iron storage 8.
Detailed study of the beneficial biochemical activities of Dps first requires its purification. Dps purification is an elaborate procedure, as Dps must be separated not only from other proteins, but also from any bound DNA 7. Our optimized purification process uses many common techniques, consisting of two ion-exchange columns and an ammonium sulfate precipitation step. Several buffer exchanges are needed, as highly concentrated Dps can precipitate out of solution in low salt conditions. Once Dps protein has been purified, it may be applied to assays that directly measure its ferroxidase activity 8, DNA-binding stoichiometry 9, and mechanisms of iron binding 10. Purified Dps also has other potential applications. The stable hollow spherical structure of Dps has been used as scaffolding for storing hydrophobic particles inside the protein cavity 11 and even as a reaction chamber to synthesize novel magnetic nanoparticles 12.
The protective ability of Dps to mediate damage due to reactive oxygen species can be clearly and directly demonstrated using the DNA protection assay 13, 14. In this in vitro procedure, radical species are produced when iron catalyzes H2O2 degradation through Fenton chemistry. These radicals directly damage DNA present in the reaction and can completely degrade it at high concentrations. Two key Dps activities may both directly counteract the effects of Fenton-mediated radical production. Dps lowers the concentration of catalytic iron through mineralization, consuming the available hydrogen peroxide in the process. Additionally, Dps binding to DNA may potentially shield it physically from radical damage and condenses it into a smaller volume with less reactive surface area. The combination of these two properties makes the DNA protection assay well suited for the purpose of measuring protective Dps activity.
The DNA protection assay is quite versatile and can be used for a variety of applications beyond Dps characterization. Radical damage is a common form of stress in cells, and many different proteins and chemicals are used to counteract it. The general principle of the assay, using DNA integrity as a marker for radical damage, can be used in combination with almost any radical-producing reaction or counteracting agent. Among others, the assay has been successfully used to determine anti-oxidative properties of K. paniculata extract for use in food industry 15, to characterize the effects of uric acid on hydroxyl damage mediation 16, and to gain new insights into the function of Fur transcriptional regulator proteins 17.
Despite the numerous uses of the assay in published papers, we found that many optimization and troubleshooting steps were required, which makes setting up the assay for the first time an unnecessarily laborious process for many researchers. The protocol we present in this article aims to remove this barrier for entry.
Protocol
1. Dps Expression and Purification
Obtaining protein of high purity is an essential first step for the DNA-protection assay. Purification of Dps protein can be performed in 4 to 5 days.
- Transform a protease-deficient strain of E. coli (such as BL21(DE3) pLysS) with a pET vector (such as pET17) into which the Dps protein-encoding sequence has been cloned.
- Streak the transformed cells out onto Luria Broth (LB) agar plates containing appropriate antibiotics (such as ampicillin and chloramphenicol for the examples provided in 1.1). Incubate the plates overnight at 37 °C.
- Inoculate a single colony from the plate into 30 ml of LB medium containing appropriate antibiotics. Incubate overnight at 37 °C while shaking at 200-250 rpm.
- Inoculate 2 x 1 L LB medium containing appropriate antibiotics with 10 ml each of the overnight culture. Grow at 37 °C, while shaking, to O.D.600 of approximately 0.6. (Growing cells to higher O.D. increases protein yield, without noticeably compromising the purity of the final product.) Induce expression of Dps by adding IPTG to a concentration of 0.3 mM, and incubate at 37 °C for 3-4 hr while shaking.
- Harvest cells by centrifugation at 6,000 x g for 15 min. Resuspend the cell pellets in 7.5 ml of DEAE buffer A (50 mM HEPES-KOH, pH 7.5, 100 mM NaCl, 0.1 mM EDTA) per L of induced cell culture. Samples may be frozen in liquid nitrogen and stored at -80 °C, if desired. The procedure may then be continued by thawing the samples in a water bath at 4 °C.
- Add a mixture of protease inhibitors (such as Calbiochem set III protease inhibitors, at 0.167 μl per ml of cell suspension) to the cell suspension to prevent degradation of overexpressed Dps.
- Prepare cell-free extract using a French press. Prime the French press with 20 ml DEAE Buffer A (if using a continuous-flow model), then disrupt the sample twice at 20 kpsi. Centrifuge the lysate at 30,000 x g for 35 min at 4 °C to clarify insoluble particles, and save the supernatant. The supernatant may then be frozen using liquid nitrogen and stored at -80 °C, if desired.
- Equilibrate a 30 ml DEAE Sepharose CL-6B column with DEAE Buffer A, using an FPLC. Run buffer through the column at 1-2 ml/min with a maximum pressure of 0.2 MPa over background. Load the cell-free extract onto the column, and begin to collect the flow-through once the O.D.280 signal begins to increase above baseline. Wash the column with DEAE Buffer A until the O.D.280 value returns to baseline, while continuously collecting the flow-through. The flow-through should contain the majority of the Dps protein, free from bound DNA. Wash the column with 100% Buffer B (50 mM HEPES-KOH, pH 7.5, 1 M NaCl, 0.1 mM EDTA). This eluate contains Dps-DNA complexes as well as other proteins and can be discarded.
- Remove additional contaminating proteins through ammonium sulfate precipitation. Determine the volume of pooled flow-through. Slowly (over a period of 10-20 min) add dry small-grain ammonium sulfate to 62% saturation (390 mg per ml of solution) at 4 °C while stirring well. Stir the mixture for an additional 20-30 min after adding the last portion of ammonium sulfate to ensure complete equilibration. Dps will remain soluble, while contaminating proteins will precipitate out of solution.
- Remove precipitated proteins by centrifugation at 20,000 x g for 30 min at 4 °C and save the supernatant.
- Slowly add an additional 227 mg ammonium sulfate per ml of supernatant to reach 94% saturation and stir for 20-30 min to ensure complete equilibration. Dps precipitates at this high salt concentration and can be collected by centrifugation at 20,000 x g for 30 min at 4 °C. Dps will be in the pellet; the supernatant can be discarded. The pellet can be stored at -80 °C if desired.
- Resuspend the Dps-containing pellet in Resuspension Buffer (50 mM HEPES-KOH, pH 7.5, 150 mM NaCl, 0.1 mM EDTA). Adjust the sample volume to 2.5 ml.
- Buffer exchange to remove the ammonium sulfate by using a PD-10 gel filtration column. Equilibrate the gel bed with 25 ml Resuspension Buffer. Apply the 2.5 ml of Dps sample to the top of the PD-10 column and allow it to soak in. Discard the flow-through. Collect Dps by eluting with 3.5 ml Resuspension Buffer.
- Centrifuge the buffer-exchanged sample at 16,000 x g for 10 min at 4 °C to remove any insoluble components, and dilute it with 6-10 ml Dilution Buffer (50 mM HEPES-KOH, pH 7.5, 0.1 mM EDTA) to ensure that the salt concentration is low enough for Dps to bind to the SP Sepharose column. Particularly concentrated samples of Dps may become less soluble upon dilution into a lower-salt buffer, as evidenced by cloudiness of the liquid, but can be completely resolubilized by the addition of a small amount of 5 M NaCl solution (an additional 10-20 mM).
- Equilibrate a 30 ml SP Sepharose Fast Flow column with SP Buffer A (50 mM HEPES-KOH, pH 7.5, 50 mM NaCl, 0.1 mM EDTA). Run buffer through the column at 1.5-2 ml/min with a pressure limit of 0.3 MPa over background. Load the sample onto the column. Wash with SP Buffer A until the O.D.280 trace returns to baseline, and discard the flow-through. Run a 150 ml linear gradient from 0 to 100% Buffer B, while collecting 2 ml fractions. Dps will elute in a sharp peak at around 50% B, though variation by 10% is possible.
- Run the elution fractions on a 15% SDS-PAGE gel, and check for DNA contamination by measuring the O.D.260/ O.D.280 ratio using a spectrophotometer. The gel should only show one band around 19 kDa, and the O.D.260/ O.D.280 is typically around 0.7. Pool the purest Dps fractions. Use a centrifugal filter unit (such as the Amicon Ultra Filtration Unit) with a 10K molecular weight cut-off to concentrate the Dps and exchange it into a storage buffer (50 mM HEPES-KOH, pH 7.5, 50 mM NaCl). Aliquot the purified Dps, and freeze in liquid nitrogen for storage at -80 °C.
2. DNA Protection Assay
The assay involves multiple time-sensitive steps and should be timed to the second to obtain reproducible results. Many ingredients and pipetting steps are involved, so a pipetting table (see Table 1) is advisable to keep track of steps and volumes. The total time required for the assay is no more than 3 hr.
- Measure 30 ml water into an airtight vial; flush the water with N2 for 10 min (using two syringe needles, one in the fluid, one above it) to remove oxygen from solution. Add 0.0168 g FeSO4•7H2O to the water to obtain a 2 mM stock solution. Seal the vial and flush with nitrogen for 5 min more, then mix. The solution must be clear; if any hint of yellow is detectable, prepare a new iron solution.
- Make a 100 mM H2O2 solution (10.87 μl of 9.2M H2O2 into 1 ml water); keep on ice and shielded from light by wrapping in aluminum foil. Use only for about 1-2 hr after preparation due to spontaneous decay. The hydrogen peroxide stock is similarly subject to breakdown, which may be partially ameliorated by proper storage (in the dark, at 4 °C). Regular checks of the stock concentration by measuring the O.D.240 are recommended.
- Thaw an aliquot of purified Dps rapidly in a room-temperature water bath, and keep on ice. Centrifuge for 10 sec at 4,000 x g in a table top picocentrifuge to precipitate aggregates. Measure concentration after centrifuging using a spectrophotometer (O.D.280=1.547 indicates a concentration of 100 μM monomer Dps).
- Dilute linear DNA from high-concentration stock using 12x reaction buffer (1 M MOPS-KOH pH 7.0, 1 M NaCl) to a concentration of 100 ng/μl. Linear DNA is used to make quantification easier, but plasmid DNA may be used as well.
- Pipette 1 μl of the DNA-buffer mixture into a PCR tube. Add enough water into the reaction so that the final reaction volume (minus SDS) will be 12 μl. This amount should be calculated in advance using the pipetting table. Add Dps to a final dodecamer concentration of 3 μM. Incubate for 15 min at room temperature to allow for Dps to bind to DNA.
- Add enough 2 mM FeSO4 solution to reach the desired concentration. A typical experiment uses a range of concentrations from 0 to 1 mM FeSO4. Higher concentrations will cause more extensive DNA degradation but may also lead to formation of ferric precipitates in the reaction mixture. Quickly add 1 μl of the 100 mM H2O2 solution (to 10 mM final concentration), and incubate at room temperature for 5 min to allow the Fenton-mediated degradation reaction to take place.
- Add 0.8 μl of 20% SDS (sodium dodecyl sulfate). Mix and incubate at 85 °C for 5 min to disrupt the Dps-DNA complexes. The SDS destabilizes the Dps-DNA complex and prevents unwanted gel shifts for the DNA, which improves the ease of quantification. An optional step is to stop the reaction by catalytically degrading the hydrogen peroxide into non-toxic products. For the conditions described in this protocol, this step is not needed to obtain a broad range of DNA degradation.
- Incubate on ice for 1 min, add loading dye, and load on an agarose gel. Run the gel, and stain for DNA using ethidium bromide. The gel should be stained post-electrophoresis rather than prior to electrophoresis, to prevent the SDS from interfering with the ethidium bromide distribution in the gel.
Representative Results
The purification process for Dps described here is very reproducible. Dps purification according to the described protocol, using 2 L of E. coli culture as a starting point, will typically yield 2.5 ml of protein containing Dps12 at concentrations between 5 and 12 μM. Longer induction times (4 hr) seem to reduce this variability. Protein purity is consistently above 99%, as evidenced by SDS-PAGE gels (Figure 1). The level of DNA contamination is consistently negligible, as evidenced both by agarose gels stained for DNA and the O.D.260/ O.D.280 ratio. This value can vary, but values are never above 0.9, indicating that DNA contamination levels remain well below 5%.
The reproducibility of the results of the DNA protection assay will depend greatly upon execution. If the concentrations of all ingredients are measured precisely (particularly the protein concentration post-centrifugation), and the incubation times for samples are consistent, the results will consistently resemble those shown in Figure 2. If conditions are less ideal, minor variability between assays will be visible, although an overall increase in DNA degradation with increasing iron concentration will still be apparent. The results obtained through the DNA protection assay can be quantified through the use of image processing software such as ImageLab. Figure 3 shows the result of this quantification, averaged over three assays. The error bars indicate the overall high level of reproducibility obtained.
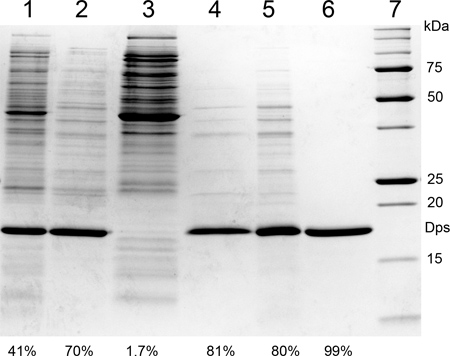
Figure 1. Intermediate steps of Dps purification, analyzed by SDS-PAGE. (1) Cell-free extract; (2) flow-through of DEAE Sepharose column; (3) eluate of DEAE column; (4) supernatant of the 60% ammonium sulfate precipitation; (5) Dps sample following the 90% ammonium sulfate precipitation and buffer exchange by PD-10 column; (6) pooled pure Dps fractions after SP Sepharose column; (7) protein molecular weight markers. The percentages on the bottom of each lane denote Dps purity as determined by image quantification software (ImageQuant TL).
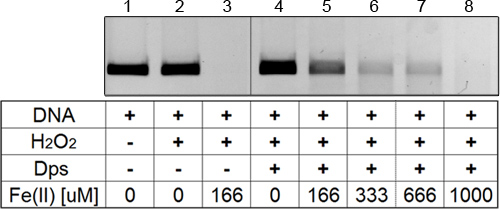
Figure 2. Dps-mediated protection of DNA from oxidative degradation. All lanes contain 100 ng of linear 5 kb DNA. (1) DNA only; (2) DNA with 1 mM H2O2; (3) DNA with 1 mM H2O2 and 166 μM FeSO4; (4) to (8) DNA with 3 μM Dps12, 1 mM H2O2, and increasing iron concentration (0 μM, 166 μM, 333 μM, 666 μM and 1,000 μM) from left to right.
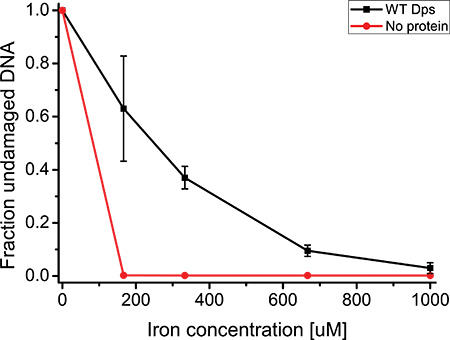
Figure 3. Fraction of undamaged DNA as a function of iron concentration, averaged over three repetitions of the DNA protection assay. Error bars denote the standard deviation of the mean. Conditions: 100 ng of 5 kb linear DNA, 1 mM H2O2, 3 μM Dps12 in 1 x reaction buffer (83 mM HEPES-KOH pH 7.0, 83 mM NaCl). The no protein control was performed using the same conditions, except with a Dps12 concentration of 0 μM.
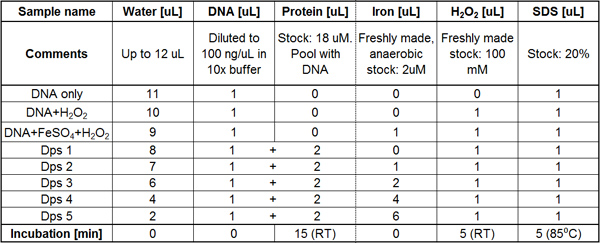
Table 1. Example of a basic pipetting table. Perform the pipetting for each column at a time, in sequential order from left to right. The incubation time following each pipetting step is indicated in the last row. The pooling of ingredients is indicated by the plus sign between cells in columns 3 and 4, indicating that the DNA and protein should be pipetted from one common tube containing both ingredients.
Discussion
The purification process of Dps as described in this article is very robust. Purity has consistently been high (> 99%); no other proteins appear on SDS-PAGE gels as visible bands. Despite this, some batches of purified Dps appear to have nuclease activity, as evidenced by partial DNA degradation when incubated with very high concentrations of Dps. This might indicate the presence of highly active DNases at low concentration that we were unable to remove through purification. However, this DNA degradation is only observed at concentrations that are not typically used for in vitro assays, so it does not usually present a problem.
The Dps purification protocol as presented in this article has several strong points. The protein does not have to be affinity-tagged, so no possibility of interfering with function exists. The purified protein is DNA-free, so in vitro assays involving DNA-binding properties will not suffer from possible contamination artifacts. Lastly, the purified Dps contains virtually no iron (<1 iron atoms/ Dps dodecamer) stored inside the protein cavity, through the ferene method of iron quantification 18, 19. This feature makes it possible to precisely control what is loaded inside the Dps cavity or to use the hollow Dps complex as a micro-reactor without worries about interference.
The DNA protection assay is a quick and reliable way to characterize the therapeutic properties of Dps in vitro. The technique offers a straightforward method to physically demonstrate the protective effect of Dps expression on cellular viability. The assay can be used to obtain quantitative data about the degree of DNA protection provided by Dps or other enzymes that counteract oxidative damage, and several steps can be taken to improve its reproducibility.
Reagent preparation is an important phase, as accurate concentration measurements are essential. It is best to use the same DNA stock for all experiments to ensure that DNA concentration does not fluctuate between measurements. In our case we extracted several milligrams of plasmid DNA using a Maxiprep kit (Promega) and digested it into linear fragments using a single-cutter restriction enzyme. Additionally, do not refreeze the Dps protein after thawing; always work with a fresh aliquot as repeated freezing and thawing may lower activity. Preparation of Dps aliquots at the time of purification in approximately the same volume as the experiment requires will prevent excessive waste of protein. Quickly centrifuging the protein before measuring concentration is crucial, as otherwise protein aggregates lead to overestimations of active protein concentration. Lastly, devoting several samples to monitor whether the reagents are working as expected (e.g. DNA alone, DNA with H2O2, DNA with H2O2 and FeSO4) is recommended for each experiment.
The DNA protection assay is quite sensitive to incubation times, so every step should be timed as accurately as possible. When doing many experiments, it is advisable to stagger samples to ensure that incubation periods remain constant between samples. Averaging over multiple experiments helps remove any errors introduced through inconsistent incubation times, but the overall error can be reduced by precise timing.
Several tips can be suggested for researchers optimizing the DNA protection assay for proteins (or chemicals) other than Dps. The most important aspect to optimize is the ratios between DNA quantity, radical production rate, and the magnitude of the protective effect that is being investigated. Until an optimal ratio was found, many assays using Dps showed either total destruction of DNA or no visible protective effect of the enzyme. In our experience, an efficient way to perform this optimization is to select a reasonably high concentration of protein, choose a DNA amount that will be clearly visible on gel without oversaturating it, and determine specific concentrations of H2O2 and FeSO4 at which all of the available DNA is destroyed. Thereafter, a series of measurements using a range of FeSO4 concentrations between zero and the determined value will reveal a dynamic range of DNA protection.
Divulgazioni
The authors have nothing to disclose.
Acknowledgements
We are grateful to Michela de Martino, Wilfred R. Hagen, and Kourosh Honarmand Ebrahimi for useful discussions. This work was supported by start-up funding from the Delft University of Technology.
Materials
| Name of Reagent/Material | Company | Catalog Number |
| BL21(DE3)pLysS competent cells | Promega | L1195 |
| pET-17b DNA | EMD-Millipore | 69663-3 |
| LB broth powder | Sigma-Aldrich | L3022 |
| Ampicillin sodium salt | Sigma-Aldrich | A0166 |
| Chloramphenicol | Sigma-Aldrich | C0378 |
| IPTG | Sigma-Aldrich | I6758 |
| HEPES | BDH | 441476L |
| Potassium hydroxide | Merck | 105033 |
| Sodium chloride | VWR | 443824T |
| EDTA | Sigma-Aldrich | E9884 |
| Protease Inhibitor Cocktail Set III | EMD-Millipore | 539134 |
| DEAE-Sepharose | Sigma-Aldrich | DFF100 |
| Ammonium sulphate | Sigma-Aldrich | A4418 |
| PD-10 Desalting Columns | GE Healthcare LS | 17-0851-01 |
| SP-Sepharose | Sigma-Aldrich | S1799 |
| Amicon Ultra Centr. Filter (10K MWCO) | Millipore | UFC901024 |
| Ferrous Sulphate heptahydrate | Sigma-Aldrich | F8048 |
| Hydrogen peroxide solution | Sigma-Aldrich | 216763 |
| MOPS | Calbiochem | 475898 |
| SDS solution | Bio-Rad | 161-0418 |
| Ethidium bromide | Sigma-Aldrich | E1510 |
| Equipment | Company | Model |
| Static incubator | Hettich | INE500 |
| Shaking Incubator | New Brunswick Sc. | Inova 44 |
| Cooled centrifuge | Beckman Coulter | Avanti J-E |
| Table-top centrifuge | Eppendorf | 5424 |
| Cell disrupter | Constant Systems Ltd. | TS2/40/AA/AA |
| FPLC Purifier | General Electric | AKTA |
| Airtight vials | Cole-Parmer | EW-08918-85 |
| Syringe needles | BD | 305128 |
| Pipettes | Eppendorf | Z683779-1EA, Z683795-1EA |
Riferimenti
- Almiron, M., Link, A. J., Furlong, D., Kolter, R. A novel DNA-binding protein with regulatory and protective roles in starved Escherichia coli. Genes Dev. 6, 2646-2654 (1992).
- Chiancone, E., Ceci, P. The multifaceted capacity of Dps proteins to combat bacterial stress conditions: Detoxification of iron and hydrogen peroxide and DNA binding. Biochimica et biophysica acta. 1800 (8), 798-805 (2010).
- Ali Azam, T., Iwata, A., Nishimura, A., Ueda, S., Ishihama, A. Growth phase-dependent variation in protein composition of the Escherichia coli nucleoid. J. Bacteriol. 181 (20), 6361-6370 (1999).
- Grainger, D. C., Goldberg, M. D., Lee, D. J., Busby, S. J. Selective repression by Fis and H-NS at the Escherichia coli dps promoter. Molecular Microbiology. 68 (6), 1366-1377 (2008).
- Martinez, A., Kolter, R. Protection of DNA during oxidative stress by the nonspecific DNA-binding protein Dps. J. Bacteriol. 179 (16), 5188-5194 (1997).
- Nair, S., Finkel, S. E. Dps protects cells against multiple stresses during stationary phase. J. Bacteriol. 186 (13), 4192-4198 (2004).
- Grant, R. A., Filman, D. J., Finkel, S. E., Kolter, R., Hogle, J. M. The crystal structure of Dps, a ferritin homolog that binds and protects DNA. Nat. Struct. Biol. 5 (4), 294-303 (1998).
- Zhao, G., Ceci, P., et al. Iron and hydrogen peroxide detoxification properties of DNA-binding protein from starved cells. A ferritin-like DNA-binding protein of Escherichia coli. J. Biol. Chem. 277 (31), 27689-27696 (2002).
- Ceci, P., Cellai, S., et al. DNA condensation and self-aggregation of Escherichia coli Dps are coupled phenomena related to the properties of the N-terminus. Nucleic Acids Res. 32 (19), 5935-5944 (2004).
- Ilari, A., Ceci, P., Ferrari, D., Rossi, G. L., Chiancone, E. Iron incorporation into Escherichia coli Dps gives rise to a ferritin-like microcrystalline core. J. Biol. Chem. 277 (40), 37619-37623 (2002).
- Swift, J., Wehbi, W. A., et al. Design of functional ferritin-like proteins with hydrophobic cavities. Journal of the American Chemical Society. 128 (20), 6611-6619 (2006).
- Ceci, P., Chiancone, E., et al. Synthesis of iron oxide nanoparticles in Listeria innocua Dps (DNA-binding protein from starved cells): a study with the wild-type protein and a catalytic centre mutant. Chimica. 16 (2), 709-717 (2010).
- Ceci, P., Ilari, A., Falvo, E., Chiancone, E. The Dps protein of Agrobacterium tumefaciens does not bind to DNA but protects it toward oxidative cleavage: x-ray crystal structure, iron binding, and hydroxyl-radical scavenging properties. The Journal of Biological Chemistry. 278 (22), 20319-20326 (2003).
- Su, M., Cavallo, S., Stefanini, S., Chiancone, E., Chasteen, N. D. The so-called Listeria innocua ferritin is a Dps protein. Iron incorporation, detoxification, and DNA protection properties. Biochimica. 44 (15), 5572-5578 (2005).
- Kumar, M. Protective effects of Koelreuteria paniculata Laxm. on oxidative stress and hydrogen peroxide-induced DNA damage. Phytopharmacology. 1 (5), 177-189 (2011).
- Stinefelt, B., Leonard, S. S., Blemings, K. P., Shi, X., Klandorf, H. Free radical scavenging, DNA protection, and inhibition of lipid peroxidation mediated by uric acid. Annals of Clinical and Laboratory Science. 35 (1), 37-45 (2005).
- Lopez-Gomollon, S., Sevilla, E., Bes, M. T., Peleato, M. L., Fillat, M. F. New insights into the role of Fur proteins: FurB (All2473) from Anabaena protects DNA and increases cell survival under oxidative stress. The Biochemical Journal. 418 (1), 201-207 (2009).
- Ebrahimi, K. H., Hagedoorn, P. L., Hagen, W. R. Inhibition and stimulation of formation of the ferroxidase center and the iron core in Pyrococcus furiosus ferritin. Journal of Biological Inorganic Chemistry. 15 (8), 1243-1253 (2010).
- Smith, F. E., Herbert, J., Gaudin, J., Hennessy, D. J., Reid, G. R. Serum iron determination using ferene triazine. Clinical Biochemistry. 17 (5), 306-310 (1984).
