Membrane Potential Dye Imaging of Ventromedial Hypothalamus Neurons From Adult Mice to Study Glucose Sensing
Summary
The activity of single neurons from adult-aged mice can be studied by dissociating neurons from specific brain regions and using fluorescent membrane potential dye imaging. By testing responses to changes in glucose, this technique can be used to study the glucose sensitivity of adult ventromedial hypothalamic neurons.
Abstract
Studies of neuronal activity are often performed using neurons from rodents less than 2 months of age due to the technical difficulties associated with increasing connective tissue and decreased neuronal viability that occur with age. Here, we describe a methodology for the dissociation of healthy hypothalamic neurons from adult-aged mice. The ability to study neurons from adult-aged mice allows the use of disease models that manifest at a later age and might be more developmentally accurate for certain studies. Fluorescence imaging of dissociated neurons can be used to study the activity of a population of neurons, as opposed to using electrophysiology to study a single neuron. This is particularly useful when studying a heterogeneous neuronal population in which the desired neuronal type is rare such as for hypothalamic glucose sensing neurons. We utilized membrane potential dye imaging of adult ventromedial hypothalamic neurons to study their responses to changes in extracellular glucose. Glucose sensing neurons are believed to play a role in central regulation of energy balance. The ability to study glucose sensing in adult rodents is particularly useful since the predominance of diseases related to dysfunctional energy balance (e.g. obesity) increase with age.
Introduction
The brain regulates energy homeostasis through the neuroendocrine and autonomic nervous systems. The ventromedial hypothalamus (VMH), comprised of the ventromedial nucleus (VMN) and the arcuate nucleus (ARC), is important for the central regulation of energy homeostasis. Specialized glucose sensing neurons, within the VMH, link neuronal activity and peripheral glucose homeostasis1. There are two types of glucose sensing neurons; glucose excited (GE) neurons increase while glucose inhibited (GI) neurons decrease their activity as extracellular glucose increases. VMH glucose sensing neurons are generally studied using electrophysiology or calcium/membrane potential sensitive dye imaging.
The electrophysiological patch clamp technique is considered to be the gold standard in the study of ex vivo neuronal activity. In this technique, a glass micropipette electrode is attached to the cell membrane via a high resistance (GΩ) seal. Patch clamp electrodes allow real time recording of action potential frequency (current clamp) or ion conductance (voltage clamp) changes within a single neuron. While the patch clamp technique provides detailed information regarding changes in specific ion channel conductances, a major drawback is that only one neuron may observed at a time. It takes approximately 30-45 min of recording to verify that one is recording from a glucose sensing neuron before even beginning a specific experimental treatment. Moreover, GI and GE neurons comprise <20% of the total VMH neuronal population. Compounding this issue is the lack, in many cases, of an identifying cellular marker for these neurons. Thus, it is clear that despite providing valuable electrical information that other techniques cannot, patch clamp analysis is laborious, time consuming and low yield.
The use of fluorescence imaging of dissociated VMH neurons allows for the study of hundreds of neurons simultaneously. Calcium sensitive dyes can be used to measure intracellular calcium changes, which indirectly correlate to changes in neuronal activity. Membrane potential sensitive dyes are used to monitor membrane potential changes. Measuring cellular membrane potential is a more direct index of neuronal activity compared to changes in intracellular calcium levels. Furthermore, membrane potential dye (MPD) imaging potentially detects smaller changes in membrane potential where action potential firing is not altered and intracellular calcium levels might not change. Both of these fluorescence imaging techniques have been used to study VMH glucose sensing neurons from juvenile mice2-7. While results are less detailed than those obtained with patch clamp electrophysiology, the strength of imaging experiments is that they simultaneously evaluate a large population of cells which inevitably include a significant number of glucose sensing neurons. MPD imaging is particularly useful for studying GI neurons which are more uniformly localized throughout the entire VMH; thus providing an adequate population to study in the dissociated VMH (~15% GI). In contrast, while GE neurons are densely localized to the ventrolateral-VMN and cell poor region between the ARC and VMN, they do not represent a significant number of neurons within the VMH (<1% GE). Moreover, by studying isolated neurons, astrocytic and presynaptic effects are eliminated. This can be an advantage in studying first order neuron effects, as well as a disadvantage since physiological connections and processes are lost.
A limiting factor in both patch clamp electrophysiology and MPD/calcium dye imaging is the need to use younger animals (e.g. mice or rats <8 weeks of age). This is predominantly due to increased connective tissue in combination with decreased neuronal viability that occurs with age. In brain-slice electrophysiology studies, increased connective tissue makes it more difficult to visualize the neurons. Increased connective tissue also makes it harder to dissociate a large number of healthy neurons for imaging studies. Furthermore, neurons from younger animals survive longer during either patch clamp recording or imaging. However, the use of young mice can be a major limitation. Neuronal activity and/or responses to neurotransmitters or circulating nutrients change with age. For example, since energy balance is closely tied to reproductive status, the hypothalamic neurons regulating energy balance may respond differently in pre- vs postpubescent animals. Additionally, many diseases require long term treatment or do not manifest until adulthood. Prime examples of such diseases are dietary obesity or Type 2 Diabetes Mellitus. Since glucose sensing neurons are believed to play a role in these diseases we developed a methodology for successfully culturing healthy adult VMH neurons for use in MPD imaging experiments.
Protocol
1. Animals
- All procedures were approved by the Institutional Animal Care and Use Committee at the University of Medicine and Dentistry of New Jersey.
- Group house male C57BL/6 mice on a 12 hr light/12 hr dark schedule and allow ad libitum access to water and food. Sacrifice at 4-5 months old. Euthanasia of the mice was performed using the surgical plane of anesthesia and a secondary form of euthanasia (i.e. penetrating incision into the chest cavity via the diaphragm). This is consistent with the AVMA Guidelines on Euthanasia.
2. Preparation of Perfusion Solution, Coverslips, Glass Pipettes, and Media
- Make 1L perfusion solution consisting of 2.5 mM KCl, 7 mM MgCl2, 1.25 mM NaH2PO4, 28 mM NaHCO3, 0.5 mM CaCl2, 7 mM glucose, 1 mM ascorbate, and 3 mM pyruvate in distilled dH2O. Adjust the osmolarity to ~300 mOsm using approximately 80 g/L sucrose. Oxygenate by bubbling with 95% O2 /5% CO2 and adjust the pH to 7.4. Each experiment will require approximately 200 ml of perfusion solution. Aliquots can be stored at -20 °C for up to 2 months.
- Make 250 ml Neurobasal stock containing Neurobasal media without glucose, 2.5 mM glucose, and 100 U/ml penicillin streptomycin. Adjust the osmolarity of the Neurobasal stock to 280 mOsm using sucrose. Make 500 ml Hibernate stock containing Hibernate-A media without glucose, 2.5 mM glucose, and 100 U/ml penicillin streptomycin.
- Stir both stocks well and filter-sterilize using Stericup vacuum filter units. Be careful not to introduce glucose or other contaminants through the glassware or stir bars. Wrap the bottles in foil to protect the media from light and store at 4 °C for up to 2 months.
- Flame the tips of 4 autoclaved glass pipettes (9 in) so that the tips are no longer sharp and the opening diameters are large (~0.9 mm), medium-large (~0.7 mm), medium (~0.5 mm), and small (~0.3 mm). The largest should be barely polished and the smallest should be about a third of that diameter.
- On the day before harvesting neurons, clean four 25 mm glass coverslips using 70% ethanol and passing over a Bunsen burner flame. Place each coverslip in a sterile 35 mm culture dish and add 2 ml sterile 1% poly-D-lysine in dH2O. Put the dishes in an incubator (37 °C) overnight. The next day, wash with sterile dH2O 3x and allow coverslips to fully dry.
- Make 75 µl aliquots of 1 M lactic acid and 200 µl aliquots of GlutaMAX; store at -20 °C. Fully thaw aliquots of lactic acid and GlutaMAX before use to ensure homogeneity.
- On the day of harvesting neurons, make 30 ml of fresh culture media consisting of Hibernate-A stock containing 1mM lactic acid, 0.5mM GlutaMAX, and 2% B27 without insulin. Vortex and adjust pH to 7.4 using 1 N NaOH.
- Put 10 ml of culture media in a 60 mm glass Petri dish on ice, put 2 ml in a 15 ml conical tube on ice and keep the remaining media at room temperature.
- On the day of harvesting neurons, make 25 ml of fresh growth media consisting of Neurobasal stock containing 1 mM lactic acid, 0.5 mM GlutaMAX, and 2% B27 without insulin. Vortex and adjust pH to 7.4 using 1 N NaOH. Allow the fresh growth media to equilibrate in an incubator (37 °C, 5% CO2) for at least 30 min.
3. Cardiac Perfusion
- Fully thaw 200 ml perfusion solution and oxygenate with 95% O2/ 5% CO2 on ice for at least 15 min.
- Wash a vibratome blade with acetone, 70% ethanol, and dH2O. Position the blade in the vibratome.
- Rinse the vibratome cooling chamber and perfusion tubing with dH2O. Fill the cooling chamber (4 °C) with perfusion solution and continue to oxygenate.
- Anesthetize a mouse with pentobarbital sodium 50 mg/ml, diluted 1:1 with sterile water, injected intraperitoneally. Verify the mouse is fully anesthetized by pinching the tail and observing no response.
- Pour cold perfusion solution into the perfusion reservoir and tubing just prior to dissection. Save 20-30 ml for brain dissection and continue to oxygenate on ice. Gravity flow from an elevated 60 ml syringe through tubing and a 20 G needle provides sufficient flow. Allow solution to run until air bubbles are washed out. Continue to oxygenate.
- Cut back skin above the ribcage of the mouse. Lift the ribcage and carefully make a horizontal cut through the diaphragm. Make two lateral cuts through the ribcage up toward the arms to expose the heart. Use forceps to hold back the ribcage.
- Make a small 2 mm cut in the right atrium to provide an exit point for blood to be washed out of the vascular system.
- Puncture the left ventricle with the perfusion needle, just enough to insert the needle opening. Hold the needle in place with one hand and start the flow of perfusion solution with the other hand. After 10-15 ml of perfusion solution, the blood should be washed out and the effluent will clear.
4. Brain Slicing and Dissection
- After cardiac perfusion, transfer oxygenated cold perfusion solution to a Petri dish on ice just prior to use.
- Quickly decapitate, remove the brain from the skull and place the brain into perfusion solution. To remove the brain: pull skin forward, make a midline cut in the skull toward the eye sockets, make 2 lateral cuts toward each eye, use forceps to pull back each side of the skull, make a coronal cut between the eye sockets, and use a spatula to gently coax the brain out into perfusion solution. Use scissors to cut the optic tracts if necessary. Do not to touch the hypothalamic area and do not pull the optic tracts because this puts too much pressure on the median eminence and underlying hypothalamic tissue.
- Use a razor blade and needle to laterally trim the brain tissue approximately 3 mm posterior and anterior to the hypothalamus while the tissue is submerged (bregma -3.52 mm). It is especially important for the anterior side cut to be level so that the brain will sit straight on the vibratome chuck.
- Remove the remaining brain tissue from solution and use filter paper to wick away solution from the mounted brain section.
- Place a dab of super glue on a vibratome chuck and spread the glue to the size of the brain. Put the brain tissue on top of the glue with the anterior side facing down and the hypothalamus facing the blade. Wick away excess glue and place the chuck into the vibratome cooling chamber. Use caution not to leave much glue as it will bubble up around the brain when placed into solution.
- With the vibratome set to a slow speed (level 2) and high amplitude (level 9), make 300-500 µm slices until reaching the hypothalamic area. Now make thin 100 µm slices cutting from the posterior to the anterior. The visual cues are as follows. Posterior to the hypothalamus the third ventricle will be very small (bregma -2.70 to -2.30 mm). At bregma -2.30 mm, the third ventricle is separated into dorsal and ventral sections on the coronal slice.
- Once sections of the third ventricle fuse, make two 500 µm slices which will contain the correct region of the VMH (bregma -2.18 to -1.22 mm). Anterior to the VMH, the third ventricle no longer extends to the ventral floor of the brain (bregma -1.06 to -0.82)8.
- Transfer these slices to the Petri dish on ice containing culture media. Dissect the VMH (VMN + ARC) as shown in Figure 1. Use a pipette to gently transfer the VMH pieces in 2 ml of culture media on ice.
5. Dissociation and Culture
- Remove the VMH from ice and place at room temperature. Add 20 U/ml papain to 4 ml of culture media. Invert to mix and place in a 34 °C water bath. Check and mix by inversion every minute until the digestion media is no longer cloudy. Filter (0.22 μm) into a sterile flask and transfer the tissue to the digestion media.
- Digest the tissue by shaking at 100 rpm at 34 °C for 30 min.
- Prepare 1 ml of media containing 8% bovine serum albumin (BSA) during the digestion. Dissolve 80 mg BSA in 1 ml of culture media and filter (0.22 μm) into a conical tube.
- Wash the tissue by transferring to 5 ml of culture media in a conical tube and slowly inverting once.
- Add 30 µl DNase enzyme to 6 ml of culture media and mix to make trituration media. Aspirate the wash media and add 3 ml of trituration media.
- Use the glass pipettes to triturate in the order of largest to smallest. Gently triturate 10x and then wait 4 min for larger pieces to settle. Use the second pipette to transfer the top 2 ml containing a dissociated cell suspension to a new conical tube. Add 2 ml of trituration media, triturate 10x and wait 3 min. Use the third pipette to transfer the top 2 ml to the cell suspension. Add 1 ml of trituration media, triturate 5x and wait 2 min. Use the fourth pipette to transfer the top 2ml to the cell suspension.
- Layer the dissociated cell suspension on top of the 8% BSA, being careful not to mix. Centrifuge at 1,000 rpm for 5 min.
- Place autoclaved 6 mm x 8 mm cloning cylinders in the center of each coverslip. Coverslips must be completely dry. Aspirate and resuspend the pellet in 440 µl warm growth media. Fill cloning cylinders with the neuronal suspension and place in the incubator for 20 min.
- Remove the cloning cylinders and very gently wash away debris. Fill each dish with 2 ml of growth media. Allow cells to recover for at least 1 hr before any assays are performed.
6. Preparation for MPD Imaging
- Make recording solution consisting of 121 mM NaCl, 4.7 mM KCl, 2 mM CaCl2, 0.1 mM MgCl2, 1.2 mM MgSO4, 0.97 mM K2HPO4, 0.23 mM KH2PO4, 5 mM NaHCO3, and 25 mM HEPES. Adjust the pH to 7.4.
- Add glucose to make the desired low glucose test solution (0.1-2.0 mM glucose). Mix thoroughly and reserve 400 ml low glucose recording solution. Add glucose to the remaining 600 ml to make 2.5 mM glucose recording solution and mix thoroughly.
- Protect dye from light as much as possible. Add 4 ml of room temperature Buffer to a Blue MPD vial. Mix thoroughly and add 5 µl dye per ml recording solution. Keep solution homogenous by mixing with pipette in between solutions. The dye contains fluorescent molecules, which enter cells with depolarization, and quencher molecules, which cannot cross the cell membrane. Aliquots may be stored at -20 °C and used within 4 days. Do not freeze-thaw more than once.
- Incubate cells in 2 ml of 2.5 mM glucose recording solution plus dye at 34 °C for 30 min. A dry heating block can be used. Protect from light.
- Prepare syringe pumps and perfusion system with 2.5 mM and 0.1 mM recording solutions plus dye. Tubing from 60 ml syringes should be connected to a manifold.
- After stopping any pump, wait for ~10 sec before switching solutions at the manifold to prevent pressure buildup in the syringe. Pressure changes alter fluid delivery to the closed chamber containing the neurons, causing changes in microscope focus during the experiment and distortion of images.
- The manifold output tubing should be connected to an in-line heater and then polyethylene tubing , which connects to a closed chamber. Bubbles should be cleared and the perfusion rate should be set to 0.5 ml/min. Protect from light.
- Transfer 25 mm coverslip with adherent neurons to the closed chamber, being careful not to allow coverslip to dry and not to introduce air bubbles. The slip is aligned in the grooves of the bottom piece, a couple drops of recording solution is placed along the sides, and the top piece is gently fitted to hold the coverslip in place.
- Use a dropper to fill the chamber with recording solution and then place a 18 mm coverslip on top. Gently fit white ring into the chamber to hold the smaller coverslip in place. Press down very slowly and lightly to prevent air bubbles from being introduced into the closed chamber.
- Start the syringe pump for the 2.5 mM glucose recording solution, press down on the white ring, and connect to the closed chamber. Slowly release pressure from white ring and connect to tubing leading to a waste container.
7. MPD Imaging
- Center the neurons using low bright field light. Try to minimize the amount of time the cells are being exposed to light.
- Allow the perfusion system to run for 10 min to allow for stabilization of temperature, focus, and dye equilibrium.
- While priming, open the MetaMorph program and take a bright field image (10X) of the neurons. Use the Auto-Focus function to take 3 stacks of fluorescent images (150 msec, Narrow Cy3 filter, 10X) and determine the focus within 5 μm. At the end of the 10 min priming period, check the focus once more.
- Begin to record fluorescent images every 30 sec for 40 min. Establish a baseline at 2.5 mM glucose for 10 min, then decrease glucose for 15 min, and finally return to 2.5 mM glucose for 15 min. Solutions are changed by stopping a syringe pump, waiting 10 sec, turning a port on the manifold, and starting another syringe pump. Changes should occur ahead of desired time points based upon the lag between the manifold and cells.
- After recording all fluorescent images, take a bright field image of the neurons.
8. MPD Imaging Analysis
- Use MetaMorph to create oval regions around each cell in the bright field image taken at the end of the experiment. Regions should be slightly larger than each neuron and can be made 1 pixel outside of the cell's dark edge. Do not choose cells that are unhealthy, overlapping, or close to debris. Unhealthy cells often appear dark. Lastly, create 5 large oval regions in areas with no cells or debris for background measurements.
- Transfer the regions to all fluorescent images and inspect to verify that no movement/shift has occurred. Use the Measure function to record the fluorescence intensity of each region for all images in an Excel file. The use of Journals in Metamorph is advised.
- In Excel, create an average of the 5 background regions for each fluorescent image. This represents the average background value at each time point. For each image/time point, subtract the background from all region measurements. The background-subtracted fluorescence intensity will be referred to as Intensity hereafter.
- For each cell, calculate the average Intensity during minutes 2-8 of recording. This represents the Baseline of the cell in 2.5 mM glucose. A 2 minute buffer on both ends of each treatment is used to minimize noise.
- For each cell, calculate the percent change from baseline: (Intensity-Baseline)/Baseline
- Create a line graph with the percent change from baseline vs time.
- Calculate the average percent change from baseline during minutes 18-23. Calculate the average percent change from baseline during minutes 35-40. While image processing is not used, noise is reduced through the averaging of intensity within each cell region, background subtraction, and the averaging of region intensity over 5 min or longer.
- Control experiments in which 2.5 mM glucose recording solution is exchanged with 2.5 mM glucose recording solution must be performed to determine the noise threshold. Calculate the average percent change from baseline during minutes 18-23 for at least 6 dishes and 3 mice. Determine the mean and standard deviation of these values.
- Set the threshold for a positive depolarization response to the mean plus 2 standard deviations. The mean plus 2 standard deviations corresponds to a difference with p<0.05. Our control experiments revealed a 10% change from baseline as the appropriate threshold value.
- For each cell, assess the reversible depolarization response based upon 2 criteria. First, the average percent change from baseline during minutes 18-23 must be greater than 10%, the threshold determined in the previous step. Second, the average percent change from baseline during minutes 35-40 must be less than half of the average percent change from baseline during minutes 18-23. The second criterion was chosen since it was found to be more rigorous than stimulation with glutamate or KCl. Unhealthy neurons often respond to stimulation with glutamate or KCl even though their responses to decreased glucose are not reversible.
- For each dish, calculate the % of depolarized neurons. This value is used to quantify the % of GI neurons among different treatment groups.
Representative Results
The precise dissection of the VMH away from other hypothalamic areas is important to obtain consistent results. The inclusion of other areas could dilute the VMH neuronal population, changing the % of depolarized neurons calculated. Furthermore, glucose sensing neurons have been identified in other hypothalamic regions, such as the lateral hypothalamus, which may differ functionally and mechanistically from VMH glucose sensing neurons. Figure 1 illustrates the correct anatomical locations for proper dissection. Following the protocol above, brain tissue containing the correct VMH region can be dissociated. Further dissection to precisely separate the VMN and ARC, while maintaining the entirety of each subpopulation, may not be possible. Figure 2 shows an example of healthy dissociated VMH neurons. Using immunocytochemistry, we confirmed that our preparation is >90% neuronal. Only healthy neurons should be used for data analysis and dishes with too many unhealthy neurons should be discarded. Neurons that are unhealthy often have very dark edges, take up the MPD to a greater extent, and have irregular shapes. Additionally, neurons should be plated at a density such that most neurons are not touching each other during recording. Neurons chosen for analysis should not be touching other cells or debris.
Data obtained from recordings of a GI neuron and a nonGI neuron are shown in Figure 3. After obtaining a baseline for 10 min, the extracellular glucose was decreased from 2.5-0.1 mM. The GI neuron shows a robust reversible response greater than the predetermined threshold of 10% change from baseline. The increase in fluorescence reflects depolarization. While GE neuron hyperpolarization responses are also detected, the frequency is too low (<1% neurons) for successful use of this technique on this subtype of glucose sensing neuron. A range of physiological glucose decreases were tested and the percentage of VMH neurons which reversibly depolarized was calculated for each dish. Figure 4 shows that a positive relationship exists between the magnitude of the glucose decrease and the percentage of depolarizing neurons. This demonstrates that successful primary culture of adult murine neurons can be used in conjunction with fluorescence imaging to allow for the study of adult VMH GI neurons.
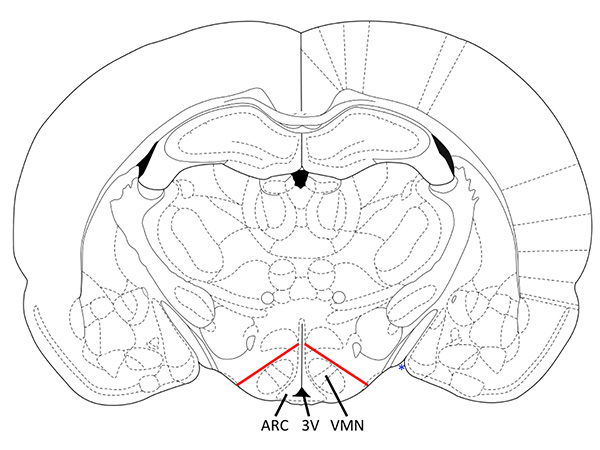
Figure 1. Coronal section with markers for correct VMH dissection. The dissected tissue contains the VMN and ARC with minimal contamination from other hypothalamic areas. Diagonal cuts should be made from ~25-30% below the top of the third ventricle to ~25-30% between the point where the cortex meets the hypothalamic area (blue *) and the third ventricle (3V). Adapted from bregma -2.8 mm Paxinos and Watson 19989, corresponding to Mouse Brain bregma -1.7mm. Click here to view larger image.
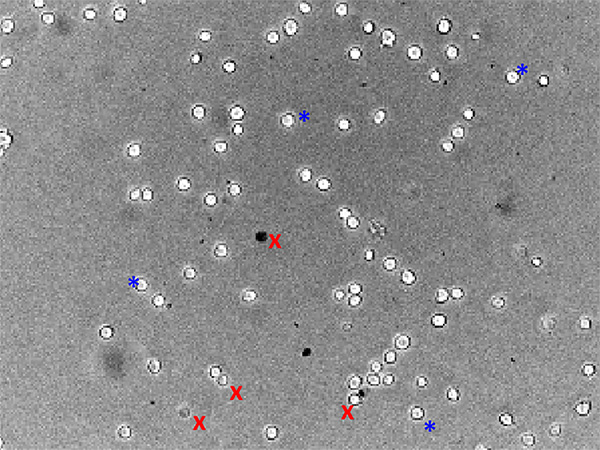
Figure 2. Brightfield image of healthy VMH neurons from an adult mouse. This image was taken 24 hr after dissociation and has been used for MPD imaging. A few examples of neurons that would (blue *) or would not (red x) be used for analysis are marked. Click here to view larger image.
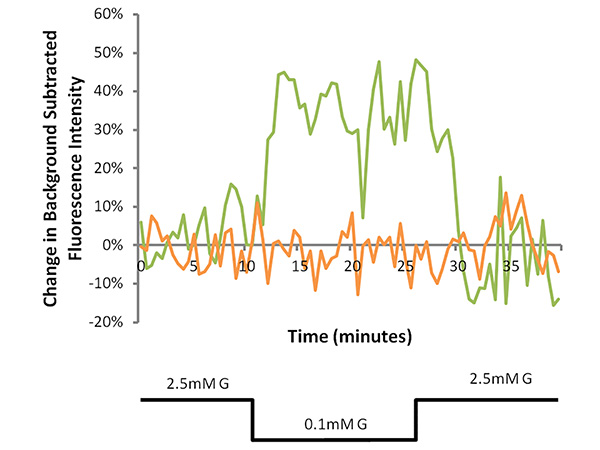
Figure 3. Representative MPD fluorescence traces of a GI neuron (green line) and a nonGI neuron (orange line). Cells were perfused with 2.5 mM glucose (G) recording solution for 10 min, followed by a decrease to 0.1 mM G for 15 min and a return to 2.5 mM G for 15 min. In the GI neuron, the percent change from baseline increased to over 10% during the 0.1 mM G perfusion period (40% average from 20-25 min) and decreased to less than half of this increase in the 2.5 mM G reversal period (15% average from 35-40 min). Click here to view larger image.
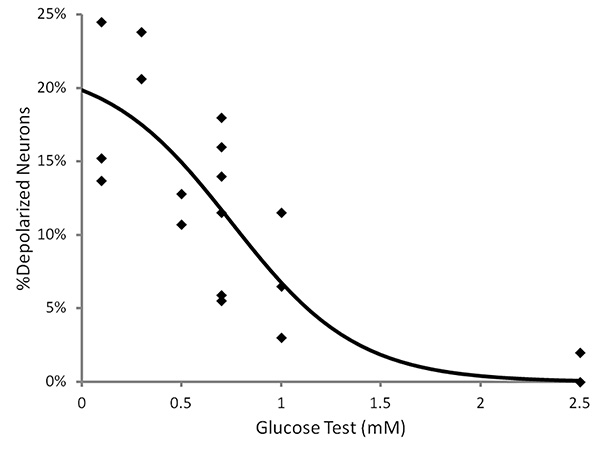
Figure 4. The percentage of adult VMH neurons which reversibly depolarize in response to decreased glucose detected using fluorescent MPD imaging. A positive relationship exists between the magnitude of the glucose decrease and the percentage of depolarizing neurons. Click here to view larger image.
Discussion
The key to being able to study activity of neurons from adult mice is the ability to dissociate healthy neurons. Dissociation of hypothalamic neurons from adult mice is more difficult at several key steps in the protocol compared to neurons from juvenile mice. We have overcome this problem in a number of ways. Making thick 500 µm brain slices minimizes mechanical damage to neurons compared to the usual 250-350 µm slices used for brain tissue from younger mice. However, thicker slices require greater attention to cardiac perfusion and papain digestion of brain tissue. If blood is seen on brain tissue, evaluate the precision and placement of the cut in the right atrium and the needle placed in the left ventricle. The flow rate and quantity of perfusion solution can also be adjusted. A thorough perfusion is imperative since blood is toxic to the neurons and will result in a lower yield of healthy neurons. Papain digestion must be efficient enough to allow for gentle trituration and easy release of single cells from brain tissue. An inefficient papain digestion is suspected when the VMH tissue sinks quickly at the end of digestion and when larger tissue pieces are present during the second trituration. With good digestion, tissue pieces are barely detected by the naked eye during the third trituration. If the trituration is not gentle, neuronal health will suffer. If an inefficient papain digestion is suspected, the concentration of papain can be increased by 5 U/ml or a new vial of papain can be used. However, the papain concentration should not be increased above 30 U/ml since this can impair neuronal health.
The exposure to BSA is another critical step in the protocol. After triturated cells have been centrifuged on the BSA gradient, both the amount of time neurons are exposed to BSA and the amount of BSA allowed to remain with the pellet should be minimized. The supernatant and the BSA gradient should be aspirated immediately following the centrifugation period. Vacuum suction can be used to remove the majority of supernatant, but the last ~100 µl should be removed manually with a pipette so that the cell pellet is not disrupted. Another important aspect of obtaining healthy neurons is to optimize cell density. Neurons plated too sparsely are not as healthy. Since so few neurons are obtained from each mouse brain, it is not practical to count cells before plating. During the time that would be spent counting, a significant number of cells would adhere to the plastic and would be lost. Using 4 dishes per mouse (~1 x 104 cells/ml) as a starting point, empirical determination of the number of dishes is the most practical approach to achieving optimal neuron density. The yield of VMH neurons from older mice can be expected to be about half of the yield from young mice. Further troubleshooting to improve neuron yield involves the quantification of neurons remaining at each step of the dissociation protocol to pinpoint steps of heavy neuronal attrition.
Using our protocol, healthy dissociated neurons can survive in culture up to 2-3 days. However, the use of filtered astrocyte-conditioned growth media allows neurons to survive up to 2 weeks in culture. Growth media can be conditioned by incubating on confluent primary astrocytes overnight. Astrocytes from the same rodent strain, sex, and brain region should be utilized. While the use of astrocyte-conditioned media introduces another variable, this modification may be necessary for alternative experiments requiring more time in culture. Moreover, for reasons which are not clear, we have found that the ability to sense glucose is easily lost with suboptimal neuronal health, despite the maintenance of responses to other stimuli, such as glutamate. Thus, additional care must be taken. To observe neuronal responses to decreased glucose, it is imperative to avoid contamination by bacteria, mold, salts, soap, and glucose. All glassware and nonsterile plastics should be thoroughly washed with dH2O. Besides living contaminants, detergents and salts would affect neuronal integrity and activity. Small amounts of glucose could significantly change glucose concentrations. This would disrupt results since glucose sensing neurons are very sensitive to small changes in extracellular glucose within the nonhyperglycemic physiological range of 0.1-2.5 mM10,11.
When utilizing MPD imaging, consistency and side-by-side experimentation between treatment groups are vital for meaningful results. Different treatment groups should be tested on the same day if possible or at least on alternating days. This minimizes possible variations in culture preparations or MPD aliquots. Additionally, we found the least variation occurs when imaging is performed within 24 hr of neuron dissociation for reasons mentioned above. Since results are analyzed as a % of depolarizing neurons, accuracy and consistency in VMH dissection and neuronal preparation is also essential. The overall VMH neuronal population harvested must be constant and must not contain neurons from other regions. Thus, the protocol above should be followed closely and precisely. However, if alternative experiments are to be performed and population maintenance is not a concern, tissue punches can be made to isolate VMN or ARC neurons.
The methods described here establish how to harvest healthy VMH neurons from an adult mouse and how to use MPD imaging to study glucose sensing neurons. Instead, experiments could be designed to examine VMH neuronal responses to stimuli other than glucose changes. Alternatively, the dissociation protocol may be extrapolated to harvest neurons from other brain regions. Using neurons from adult mice, studies can now be performed using murine disease models that develop or progress with age. Moreover, healthy adult neurons can be further studied using techniques other than MPD imaging such as calcium imaging, electrophysiology, single-cell PCR, immunocytochemistry or a combination of these techniques.
Divulgazioni
The authors have nothing to disclose.
Acknowledgements
NIH R01 DK55619, NIH R21 CA139063
Materials
| Neurobasal-A Medium (Custom) | Invitrogen | 0050128DJ | custom made glucose free |
| Hibernate-A Medium (Custom) | BrainBits | custom made glucose free | |
| Penicillin streptomycin (20,000 U/ml) | Invitrogen | 15140 | other vendors acceptable |
| Stericup vacuum filter units (0.22 μm) | Millipore | other vendors acceptable | |
| 25 mm Glass coverslips | Warner | #1 25mm round | |
| 18 mm Glass coverslips | Warner | #1 18mm round | |
| GlutaMAX | Invitrogen | 35050 | |
| B27 minus insulin (50x) | Invitrogen | 0050129SA | |
| Razor blade | VWR | 55411 | |
| Vibratome & cooling chamber | Vibratome | Series 1000 Sectioning system | |
| Vibratome blades | Polysciences | 22370 | injector or double edge blades from other vendors acceptable |
| Papain, suspension | Worthington | LS003124 | |
| BSA, suitable for cell culture | Sigma | other vendor acceptable | |
| DNAse, for cell culture | Invitrogen | other vendor acceptable | |
| cloning cylinders, 6 mm x 8 mm | Bellco Glass | 2090-00608 | |
| Membrane Potential Dye (blue) | Molecular Devices | R8042 | |
| In-line heater | Warner | SF-28 | |
| Syringe pumps | WPI | sp100i | other vendor acceptable |
| Closed chamber | Warner | RC-43C | |
| Polyethylene tubing | Warner | PE-90 | |
| Metamorph | Molecular Devices | alternate image analysis software acceptable | |
| Microscope | Olympus | BX61 WI |
used with 10X objective |
| Camera | Photometrics | Cool Snap HQ | |
| Narrow Cy3 Filter Set | Chroma | 41007a | |
| Illumination System | Sutter Instruments | Lambda DG-4 |
Riferimenti
- Routh, V. H. Glucose-sensing neurons: are they physiologically relevant?. Physiol. Behav. 76, 403-413 (2002).
- Canabal, D. D., Potian, J. G., Duran, R. G., McArdle, J. J., Routh, V. H. Hyperglycemia impairs glucose and insulin regulation of nitric oxide production in glucose-inhibited neurons in the ventromedial hypothalamus. Am. J. Physiol. 293, 592-600 (2007).
- Canabal, D. D., et al. Glucose, insulin, and leptin signaling pathways modulate nitric oxide synthesis in glucose-inhibited neurons in the ventromedial hypothalamus. American journal of physiology. Reg. Integr. Comp. Physiol. 292, 1418-1428 (2007).
- Murphy, B. A., Fakira, K. A., Song, Z., Beuve, A., Routh, V. H. AMP-activated protein kinase and nitric oxide regulate the glucose sensitivity of ventromedial hypothalamic glucose-inhibited neurons. Am. J. Physiol. Cell Physiol. 297, C750-C758 (2009).
- Murphy, B. A., et al. Fasting enhances the response of arcuate neuropeptide Y-glucose-inhibited neurons to decreased extracellular glucose. Am. J. Physiol. Cell Physiol. 296, C746-C756 (2009).
- Kang, L., et al. Glucokinase is a critical regulator of ventromedial hypothalamic neuronal glucosensing. Diabetes. 55, 412-420 (2006).
- Kang, L., et al. Prior hypoglycemia enhances glucose responsiveness in some ventromedial hypothalamic glucosensing neurons. Reg. Integr. Comp. Physiol. 294, R784-R792 (2008).
- Paxinos, G., Franklin, K. B. J. . The Mouse Brain in Stereotaxic Coordinates. , (2004).
- Paxinos, G., Watson, C. . The Rat Brain in Stereotaxic Coordinates. , (1998).
- Song, Z., Levin, B. E., McArdle, J. J., Bakhos, N., Routh, V. H. Convergence of pre- and postsynaptic influences on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes. 50, 2673-2681 (2001).
- Song, Z., Routh, V. H. Differential effects of glucose and lactate on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes. 54, 15-22 (2005).
