Figure 1 provides an overview of ERRBS, highlighting key steps, which are explained throughout the protocol described. ERRBS libraries were prepared using 50 ng input DNA.
Evaluate the quality of the libraries prepared. Library production routinely yields fraction sizes of 150-250 bp and 250-400 bp (Figure 3A-C). Slight differences in library size distributions between samples are expected. Note that in both lower and higher library fractions there are very intense DNA sizes, indicative of enrichment of a particular sequence. MspI digestion results in the enrichment of a family of repetitive DNA sequences present in the human genome at 190 bp, 250 bp and 310 bp in the ERRBS libraries. These three repeats represent a characteristic signature of an ERRBS library20 (see Figures 3A-C and 3G). Representative libraries were sequenced on a next-generation sequencer using single-end reads. When loading at the recommended library concentration on an Illumina HiSeq 2500 sequencer, cluster densities of 500,000-700,000 per mm2 are expected. At this clustering density, 81.6% ± 3.14% (n = 81) of the clusters pass filter (Figure 4A). Due to the low complexity end of the library inserts (MspI recognition site: C^CGG), intensity values and quality scores recorded during sequencing are highly variable in the first three bases (Figure 4B-C), however, if an independent control lane is included (see discussion), 85% of bases will have quality scores of 30 or greater (Q30 values; Figure 4D).
Data alignment and cytosine methylation determination as described in the protocol yields base-pair resolution data (Table 7). For the human genome, a 51-cycle single-read sequencing run of an ERRBS library in one lane of a HiSeq 2500 in high output mode regularly generates 153,194,882 ± 12,918,302 total reads that after quality filtering and adapter trimming yields 152,231,183 ± 13,189,678 reads for input into the analysis pipeline. Average mapping efficiency for an ERRBS library is typically 62.95% ± 5.92% with representation of 3,183,594 ± 713,547 CpGs with a minimum coverage per CpG of 10x and an average coverage per CpG of 84.94 ± 16.29 (n = 100).
The ERRBS protocol is amenable to multiplexing (see Supplemental file 1: Protocol adaptation for multiplexed sequencing). Data from representative sequencing runs is summarized in Figure 5. Data from multiplexed sequencing runs (51-cycle single-read sequencing run; n = 128 for two libraries per lane; n = 11 for three libraries per lane; n = 11 for four libraries per lane) were compared to a full lane sequencing of an ERRBS library (51-cycle single-read sequencing runs; n = 100) as well as downsampling a single lane to simulate 50%, 33% and 25% of reads per lane (2, 3, and 4 sample multiplexing per lane respectively; n = 3). As the number of reads per sample decreases with the multiplexing factor, the number of CpGs covered at a minimum coverage of 10x and the coverage per CpG decreases as well (Figure 5 and Table 8). Mean conversion rates of non-CpG sites expected are 99.85% ± 0.04% (n = 400). Conversion rates lower than 99% may indicate less than optimal bisulfite conversion that can result in high rates of false methylation levels.
Data from an ERRBS library prepared from a representative human genomic DNA was analyzed in R 2.15.245 using the methylKit package26 (see Supplemental code file 1 for command details). The data can be visualized in commonly used genome browsers (Figure 6A). The cytosine methylation data is equally derived from both strands (Figure 6B) and ranges the entire spectrum of potential cytosine methylation levels (Figure 6C). Analysis of technical replicates from a representative human DNA sample yields high concordance between the data results (Figure 6D) and covers CpGs in a broad spectrum of genomic loci (Figure 6E and F and as previously described26). While technical replicas will yield high R2 values (greater than 97%), biological replicas will yield R2 values ranging from 0.92 to 0.9626, and comparing different human cell types will yield R2 values lower than 0.86 (data not shown).
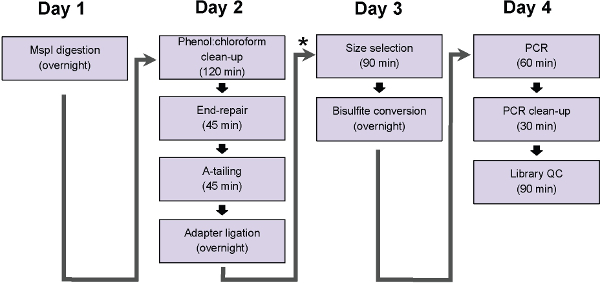
Figure 1: Flow chart of the ERRBS protocol steps. Chart represents steps, which can be completed in a traditional work day. * indicates a potential pause point (immediately following ligation clean up and before size selection, protocol step 5) at which samples can be frozen at -20 °C before proceeding with the duration of protocol.
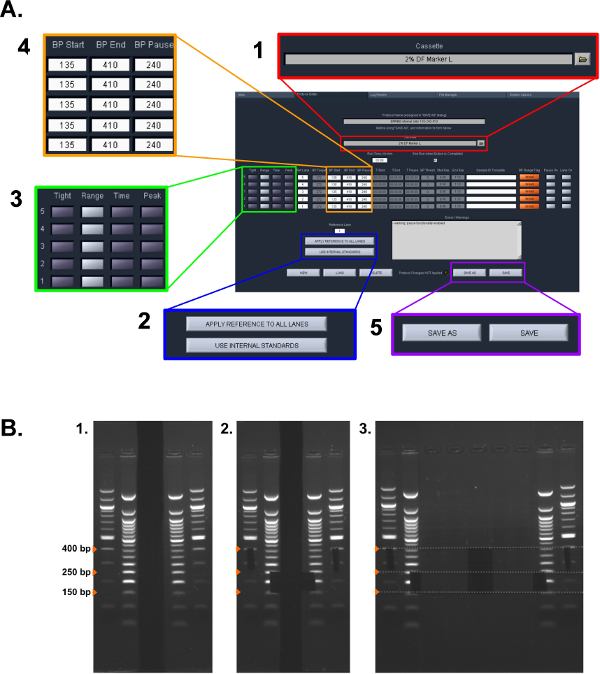
Figure 2: Size selection protocol. (A) Screen shot of settings used in the ERRBS Pippin Prep protocol (see protocol section 5.1.2 – 5.1.6): (1) Select Cassette type. (2) Select standard to be used. (3) Select the collection mode for each lane. (4) Enter the collection bp ranges. (5) Save the protocol. (B) Stages of the manual gel extraction used in protocol section 5.2: (1) Visualized gel ladders. (2) Marked Sizes for size selection using a razor blade. (3) Image of excised samples (lower fraction: 150-250 bp and higher fraction: 250-400 bp). Please click here to view a larger version of this figure.
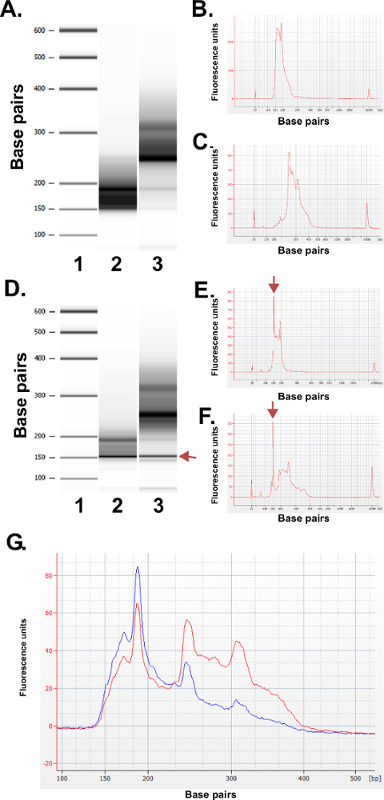
Figure 3: Quality control results for representative ERRBS libraries prepared from human DNA samples using a bioanalyzer machine. (A) Gel-like image showing a standard ladder (1), lower library fraction (135-240 bp fraction from Pippin Prep); 2) and the higher library fraction (240-410 bp fraction from Pippin Prep); 3). (B) Bioanalyzer electropherogram of the expected lower library fraction. (C) Bioanalyzer electropherogram of the expected higher library fraction. D–F) Representative data from a poor quality library prep. Gel-like image (D) of the standard ladder (1), lower library fraction (2) and the higher library fraction (3). The band at 150 bp marked with an arrow indicates excessive amounts of adapter. Electropherogram of the lower (E) and higher library fractions (F) with the excess adapter peaks at 150 bp (marked with arrows). (G) Bioanalyzer electropherogram of a pooled ERRBS library for sequencing. Red trace represents a high quality pooled library with equal representation of higher and lower fractions. Blue trace represents a pooled library not adequate for sequencing due to a lack of equal representation of the higher and lower fractions. Please click here to view a larger version of this figure.
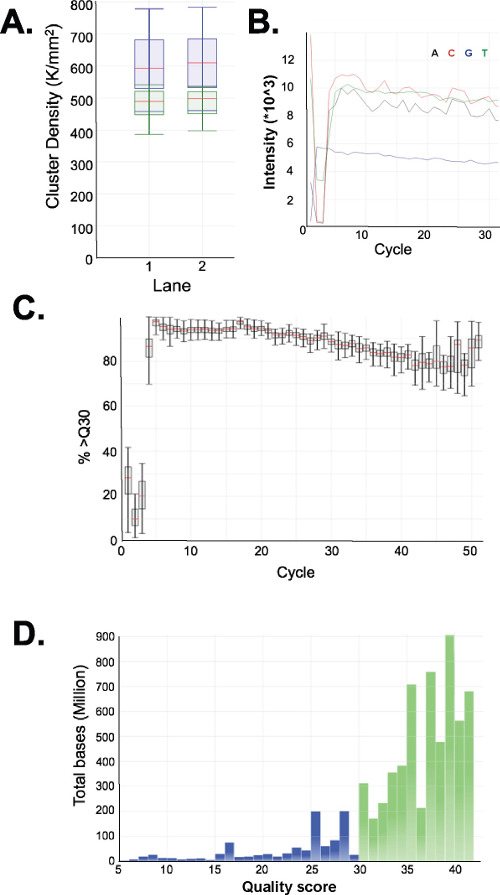
Figure 4: Sequencing charts for a representative ERRBS 51-cycle single-read sequencing run on a HiSeq 2500 sequencer in high output mode. (A) Cluster densities (K/mm2 = 1,000 clusters per millimeter squared; blue) and cluster densities passing filter (green) in two lanes with ERRBS libraries. (B) Typical intensities seen in the first 30 cycles in a lane with an ERRBS library. Note the CGG signature from MspI digestion in the intensities of the first three cycles. (C) Percentage of bases with a quality score of 30 or higher (%>Q30) for each cycle in one ERRBS lane. (D) Quality score distribution for all cycles in one ERRBS lane. Blue = less than Q30, Green = greater than or equal to Q30. In this lane, 84.7% of bases had quality scores of 30 or higher.
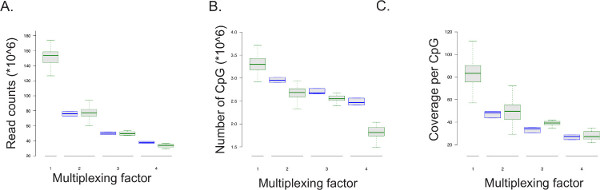
Figure 5: Sequencing output results. Box plots of experimental data from multiplexed and single sample per lane sequencing runs (displayed as green boxes) and of data derived by simulated downsampling from sequencing runs of three ERRBS libraries (displayed as blue boxes; sampled five times for each sequencing run) from 51-cycle single-read sequencing runs. The multiplexing factor corresponds to the number of ERRBS libraries sequenced per lane. 1 = whole lane or 100% of reads and represents data from a single ERRBS library per lane; 2 = 50% of lane and represents data from two ERRBS libraries per lane; 3 = 33% of a lane and represents data from three ERRBS libraries per lane; and, 4 = 25% of a lane and represents data from four ERRBS libraries per lane. (A) The read counts, or number of sequences analyzed, per multiplexing factor. (B) The number of CpG’s covered by the sequencing data per multiplexing factor. (C) The mean coverage per CpG per multiplexing factor. Please click here to view a larger version of this figure.
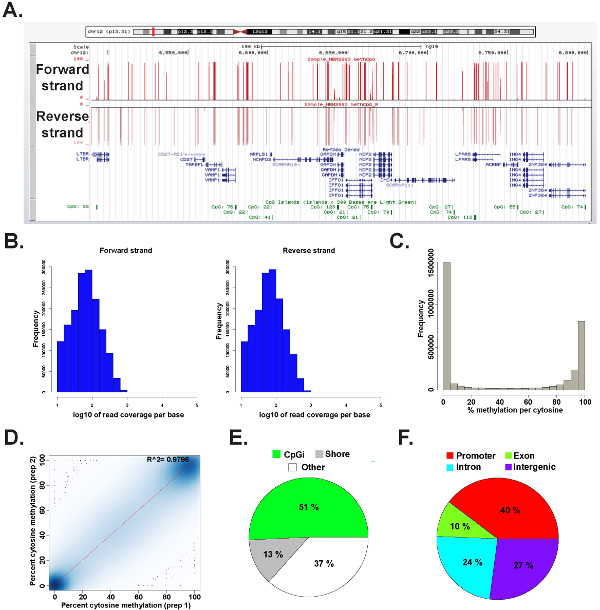
Figure 6: Representative data from an ERRBS library prepared from human genomic DNA. (A) University of California, Santa Cruz (UCSC) genome browser43 image of representative data from an ERRBS sequencing lane. The y-axis scale bar represents 0-100% methylation at each cytosine covered with a minimum of 10x. The top custom track represents the forward strand and the lower custom track represents the reverse strand. Shown is chr12:6,489,523-6,802,422 (hg19) inclusive of refseq genes and CpG islands within this genomic region. (B) Distribution histograms of CpG coverage along forward and reverse strands in a representative human CD34+ bone marrow sample. (C) Distribution histogram of CpG methylation levels along both strands in a representative human CD34+ bone marrow sample. (D) Correlation plot of CpG methylation levels from a representative technical replica of a human DNA sample. (E) Pie chart illustrating the proportions of CpGs covered in ERRBS which annotated to CpG islands (light green), CpG shores (gray) and other regions (white) in a representative sample prepared from human genomic DNA. (F) Pie chart illustrating the proportions of CpGs covered in ERRBS which annotated to gene promoters (red), exons (green), introns (blue) and intergenic regions (purple). Please click here to view a larger version of this figure.
| Reagent | Volume | Comment |
| 10x T4 DNA Ligase Reaction Buffer | 10 µl | |
| Deoxynucleotide triphosphate (dNTP) Solution Mix | 4 µl | mix of 10 mM of each nucleotide |
| T4 DNA Polymerase | 5 µl | 3,000 units/ml |
| DNA Polymerase I Large (Klenow) Fragment | 1 µl | 5,000 units/ml |
| T4 Polynucleotide Kinase | 5 µl | 10,000 units/ml |
| DNase-free water | 45 µl |
Table 1: End repair reaction reagents. Reagent names and quantities used in the end repair reaction (protocol step 2.1).
| Reagent | Volume | Comment |
| 10x reaction buffer | 5 µl | for example, NEBuffer 2 |
| 1 mM 2'-deoxyadenosine 5'-triphosphate (dATP) | 10 µl | |
| Klenow Fragment (3’→5’ exo-) | 3 µl | 5,000 units/ml |
Table 2: A-tailing reaction reagents. Reagent names and quantities used in the A-tailing reaction (protocol step 3.1).
| Reagent | Volume | Comment |
| 15 µM annealed adapters in DNase-free water | 3 µl | PE adapter 1.0 and PE adapter 2.0; see Table 4 for sequences and reference |
| 10x T4 DNA Ligase Reaction Buffer | 5 µl | |
| T4 DNA Ligase | 1 µl | 2,000,000 units/ml |
| DNase-free water | 31 µl |
Table 3: Adapter ligation reaction reagents. Reagent names and quantities used in the adapter ligation reaction (protocol step 4.2).

Table 4: Oligos used in the ERRBS protocol. List of oligos used throughout the ERRBS protocol in the ligation reaction (protocol step 4) and PCR amplification steps (protocol step 7). Please click here to view a larger version of this table.
| Reagent | Volume | Comment |
| 10x FastStart High Fidelity Reaction Buffer with 18 mM magnesium chloride | 20 µl | |
| 10 mM dNTP Solution Mix | 5 µl | |
| 25 µM PCR PE primer 1.0 | 4 µl | See Table 4 |
| 25 µM PCR PE primer 2.0 | 4 µl | See Table 4 |
| FastStart High Fidelity Enzyme | 2 µl | 5 units/µl FastStart Taq DNA Polymerase |
| DNase-free water | 125 µl |
Table 4: Oligos used in the ERRBS protocol. List of oligos used throughout the ERRBS protocol in the ligation reaction (protocol step 4) and PCR amplification steps (protocol step 7).
| Reagent | Volume | Comment |
| 10x FastStart High Fidelity Reaction Buffer with 18 mM magnesium chloride | 20 µl | |
| 10 mM dNTP Solution Mix | 5 µl | |
| 25 µM PCR PE primer 1.0 | 4 µl | See Table 4 |
| 25 µM PCR PE primer 2.0 | 4 µl | See Table 4 |
| FastStart High Fidelity Enzyme | 2 µl | 5 units/µl FastStart Taq DNA Polymerase |
| DNase-free water | 125 µl |
Table 5: PCR reaction reagents. Reagent names and quantities used in the PCR amplification reaction (protocol step 7.1).
| Protocol step | Reagent/protocol detail | Input DNA amount | ||
| 5-10 ng | 25 ng | 50 ng | ||
| 1 | MspI enzyme | 1 µl | 2 µl | 2 µl |
| MspI digest reaction volume | 50 | 100 | 100 | |
| 4 | Adapters in ligation reaction | 1 µl | 2 µl | 3 µl |
| Ligation reaction volume | 20 µl | 25 µl | 50 µl | |
| 5 | Size selection protocol | Manual gel only | Pippin Prep or manual gel | Pippin Prep or manual gel |
| 7 | PCR primer concentration | 25 µM | 25 µM | 10 µM for 14 cycles; 25 µM for 18 cycles |
| Number of PCR cycles | 18 | 18 | 14-18 | |
Table 6: Protocol step modifications for input material quantities ranging from 5-50 ng. Several steps throughout the protocol require modification of reagent quantities used to generate high quality libraries from various quantities of starting materials. Changes to key reagent quantities are included here. Adjust buffer and water volumes in reactions accordingly.
| Chr | Base | Strand | Coverage | freqC | freqT |
| chr1 | 10564 | R | 366 | 85.52 | 14.48 |
| chr1 | 10571 | F | 423 | 91.25 | 8.75 |
| chr1 | 10542 | F | 432 | 91.2 | 8.8 |
| chr1 | 10563 | F | 429 | 94.64 | 5.36 |
| chr1 | 10572 | R | 366 | 96.99 | 3.01 |
| chr1 | 10590 | R | 370 | 88.11 | 11.89 |
| chr1 | 10526 | R | 350 | 92 | 8 |
| chr1 | 10543 | R | 368 | 92.93 | 7.07 |
| chr1 | 10525 | F | 433 | 91.92 | 8.08 |
| chr1 | 10497 | F | 435 | 88.74 | 11.26 |
Table 7: Representative ERRBS data. After data alignment and cytosine methylation determination, base pair data is obtained. For each CpG covered, the alignment protocol as described will determine the genomic coordinate (columns: chr = chromosome, Base and Strand), the coverage rate of the specific locus (Coverage), and the rate of detection cytosine versus thymidine as percent (freqC and freqT respectively).
| Number of ERRBS libraries per lane | Mean number of uniquely aligned reads | Mean number of CpGs covered | Mean coverage per CpG |
| 1 | 152,231,184 ± 13,189,678 | 3,183,594 ± 713,547 | 85 ± 16 |
| 2 | 77,680,837 ± 7,657,058 | 2,674,823 ± 153,494 | 49 ± 9 |
| 3 | 49,938,156 ± 2,436,865 | 2,552,186 ±- 76,624 | 39 ± 2 |
| 4 | 34,457,208 ± 4,441,686 | 1,814,461 ± 144,339 | 28 ± 4 |
Table 8: Representative parameters from sequencing single and multiplexed ERRBS libraries. Shown is data per lane from 51-cycle single-read sequencing runs: mean and standard deviations of uniquely aligned reads, number of CpGs covered and coverage per CpG site obtained from sequencing single ERRBS libraries per lane (n = 100), two ERRBS libraries per lane (n = 128), three ERRBS libraries per lane (n = 11), and four ERRBS libraries per lane (n = 11).
