Note: All cells were isolated from resected non-tumorous human liver tissue, which remained after partial liver resection with primary or secondary liver tumors. Informed consent of the patients was obtained according to the ethical guidelines of the Charité – Universitätsmedizin Berlin.
1. Preparation of Materials and Solutions
- Sterilize all instruments and the materials in advance to avoid bacterial contamination during the isolation process.
- Prepare the solutions required for the perfusion of the liver tissue sample, the isolation process of hepatocytes and non-parenchymal liver cells and the cultivation of primary human liver cells according to the Tables 1 and 2, with exception of Digestion-Solution which is prepared freshly prior to use. All solutions can be stored at 4 °C and it is recommended to use them within 4 weeks after preparation.
- Sterilize all solutions using a 0.22 µm bottle top filter.
2. Preparation of Perfusion Equipment
- Set up the equipment for the perfusion and digestion of the liver tissue sample as shown in Figure 1A.
- Adjust the water bath temperature to 39 °C to ensure optimal collagenase P activity during the perfusion and digestion.
3. Perfusion and Digestion of the Liver Tissue Sample (1.5 hr)
- Select a tissue sample with an intact Glisson's capsule from the resected liver tissue. When cutting the tissue sample, try to obtain a small cutting surface with good visible vessels. Avoid warm ischemia times by transporting and handling the liver tissue sample on ice until perfusion.
- Take the tissue weight under sterile conditions and place the liver tissue sample in a Petri dish in the laminar air flow. Clean the surface of the tissue sample with a sterile compress from remaining blood and flush the cannula set using 1x Perfusion-Solution I to ensure that all cannula were permeable.
- Use tissue glue to fix the olives of the cannulas in some larger blood vessels. Depending on the size of the liver tissue sample and the number of the vessels on the surface, use a cannula set with 3 to 8 cannulas. Test the perfusion and check for leakages. Close all blood vessels, which leak clear 1x Perfusion-Solution I, with tissue glue.
- Place the cannulated liver tissue sample into the Büchner funnel on its perforated filter disc (Figure 1A).
- Set the flow rate of the peristaltic pump between 7.5 ml/min and 14.6 ml/min depending on the number of cannulas used and on the resistance of the liver tissue. Adjust the flow rate each time to ensure that there is a current but slow perfusion. Perfuse the tissue until the whole blood is flushed out but at least 20 min. Observe the tissue become brighter in areas with good perfusion.
Note: In some cases, it may be necessary to clamp one of the cannulas with plastic clamps or to increase the inner pressure of an area by pushing softly with a spatula against the liver capsule, to optimize the perfusion. A complete color change to a light yellow to light brownish color indicates a good perfusion. - Change the perfusion fluid to Digestion-Solution containing collagenase P (Table 1).
- Rearrange the setup (Figure 1A) for the digestion step. Therefore perform a circular flow of Digestion-Solution according to Figure 1B for up to 15 min.
Note: It is critical to stop the perfusion immediately when the liver tissue sample is sufficiently digested. A good digestion can be observed, when the tissue shows no sign of elasticity as assessed by maintenance of capsule deformations, when it is pushed with a spatula.
4. Isolation of Hepatocytes (1 hr)
- Turn the peristaltic pump off and place the liver tissue sample in a glass dish. Rinse the outside of the tissue sample with ice cold Stop-Solution (Table 1). Remove the cannulas from the liver tissue sample. Use a scalpel to open the liver tissue sample, by incising in the middle of the area where the cannulas were attached. Keep care that the Glisson´s capsule stays intact.
- Rinse the inside of the tissue sample and then cover the whole tissue sample with ice cold Stop-Solution. Shake the tissue gently to release the cells out of the tissue.
- Collect the cell suspension and filter it through a gaze funnel (plastic funnel lined with gauze compress) into 50 ml plastic tubes. Add more Stop-Solution to the liver tissue sample until a final volume of 500 ml is consumed.
- Centrifuge the cell suspension at 50 x g, 5 min, 4 °C. Collect the supernatant for later non-parenchymal cell isolation. Wash the cell pellet with PBS (Figure 2A).
- Centrifuge the cell suspension again at 50 x g, 5 min, 4 °C. Collect the supernatant and re-suspend the pellet in Hepatocyte Incubation medium (Table 2, Figure 2B).
- Determine the cell number and viability in the resulting cell suspension using trypan blue staining. Count the living and dead cells in a Neubauer counting chamber. Calculate the cell number, viability and yield of PHH using the formulas below.
yield (counted cells) = counted cells x dilution factor x volume of cell suspension (ml) x 10,000
yield (hepatocytes/(g liver tissue sample)) = (yield (hepatocytes/(ml media)) x Volume of cell suspension (ml))/(weight of liver tissue sample (g))
viability (%) = 100% x (number of live cells)/(total cell number)
5. Purification of Hepatocytes (1 hr)
Note: This purification step is recommended, if the viability is lower than 70%.
- Perform all steps on ice. Prepare a 25% density gradient by mixing 5 ml density gradient solution and 15 ml PBS for density gradient centrifugation.
- Put a maximum of 50 Mio cells in total out of the hepatocyte rich cell suspension carefully and slowly on top of the 25% density gradient layer to ensure that a clear separation of both layers is achieved (Figure 2C). Put the tubes carefully into the centrifuge and centrifuge at 1,250 x g, 20 min, 4 °C without brake (Figure 2D).
- Aspirate the remaining cell suspension and the dead cells in the interphase. Depending on the fat content one might also aspirate the density gradient-solution.
Note: PHH with low lipid content form a dense pellet and the density gradient can be aspirated completely. PHH with high lipid content form a more diffuse pellet and a lot of viable cells may remain in the density gradient solution above the pellet. - Re-suspend the hepatocyte pellets with PBS and centrifuge again at 50 x g, 5 min, 4 °C. Pool the pellets, wash again with PBS and re-suspend purified PHH in Hepatocyte Incubation medium. Perform cell counting as described in step 4.6.
6. Cultivation of Hepatocytes
- Prepare the cell culture dishes for seeding of PHH by coating them with an extracellular matrix, for example rat tail collagen (collagen type I). Prepare the rat tail collagen according to the protocol established by Rajan et al.17
- Dilute the rat tail collagen stock solution 1:200 in PBS. Transfer 100 µl/cm2 rat tail collagen solution into the culture dishes, taking care that the whole surface is covered. Incubate the cell culture plastics for 20 min at room temperature. Aspirate the remaining rat tail collagen solution.
- Seed 15 x 104 hepatocytes/cm2 in Hepatocyte Incubation medium on culture dishes coated with rat tail collagen. Cultivate the cells in a humidified incubator at 37 °C, 5% CO2 for at least 4 hr. After 4 hr the hepatocytes have adhered and the medium can be changed.
- Perform investigations depending on the experimental setup. A culture time of 48 hr is recommended to allow the cells to recover from the isolation process.
7. Isolation of Non-parenchymal Liver cells (1.5-2 hr)
- Centrifuge the collected supernatant (step 4.5 and 4.6) at 72 x g, 5 min, 4 °C to eliminate the remaining erythrocytes and hepatocytes. Pool the supernatants and centrifuge them twice to gain two cell pellets: 300 x g, 5 min, 4 °C for the sedimentation of HSC, LEC and partly KC and 650 x g, 7 min, 4 °C for sedimentation of the remaining KC.
- Pool both pellets and re-suspend them in HBSS. Prepare a 25% and a 50% density gradients by mixing density gradient solution and PBS for density gradient centrifugation (25% density gradient solution: 5 ml density gradient solution and 15 ml PBS, 50% density gradient solution: 10 ml density gradient solution and 10 ml PBS, see Figure 2). Place the 25% density gradient solution carefully on top of the 50% density gradient solution layer.
- Put the NPC suspension carefully and slowly on top of the 25% density gradient solution layer in a way that a clear separation of both layers is achieved.
- Centrifuge the cell suspension on the density gradient at 1,800 x g, 20 min, 4 °C without brake (Figure 2.2).
- Aspirate dead cells and cell debris from the uppermost layer. The NPC are located in the interphase between the 25% and 50% density gradient layer (Figure 2). Collect NPC, wash them with HBSS and centrifuge the cell suspension applying the above described dual centrifugation step (step 7.2.).
8. Separation of Kupffer Cells (Adherence Separation Step) (1 hr)
- Perform a cell count for the KC in the NPC fraction as described in step 4.6. (For appearance of KC in suspension see Figure 3B). Centrifuge the NPC fraction with the above described dual centrifugation step (step 7.2) and re-suspend the NPC in Kupffer Cell seeding medium (Table 2).
- Seed the KC containing fraction on plastic cell culture vessels at a density of 5 x 105 KC/cm2. Incubate the KC cultures for 20 min in a humidified incubator at 37 °C, 5% CO2. Primary KC adhere on cell culture plastics within a short period of time (Figure 2.3).
- Collect the supernatant containing not adhered NPC, consisting mainly of LEC and HSC. Pool the supernatants for later separation of LEC (see section 9) and HSC (see section 10). Wash the adherent KC with HBSS and cultivate them in Kupffer Cell culture medium (Table 2) at 37 °C, 5% CO2 in a humidified incubator.
9. Separation of Endothelial Cells (1.5 hr)
- Centrifuge the collected supernatant (step 8.5.) at 300 x g, 5 min, 4 °C. Wash the pellet with PBS. After centrifugation at 300 x g, 5 min, 4 °C re-suspend the cells in Stellate Cell/Endothelial Cell separation medium and perform a cell count for all remaining cells as described in step 4.6.
- Re-suspend 1 x 107 Mio cells in 1 ml Stellate Cell/Endothelial Cell separation medium, add 20 µl Blocking Solution from the MACS-KIT and 20 µl of the CD31 Micro Beads for immunolabeling and incubate the resulting suspension for 15 min at 4 °C temperature (Figure 2.4).
- Separate LEC from HSC as described in the manufacturer´s protocol for the magnetically activated cell sorting system MACS (Figure 2.5). Elute magnetically retained CD31-positive LEC and suspend them in Stellate Cell/Endothelial Cell culture medium (Table 2).
- Perform cell counting for LEC as described in step 4.6. Seed LEC in a density of 1.25 x 105 cells/cm2 in cell culture vessels coated with rat tail collagen (see step 6.1). Cultivate the cells at 37 °C, 5% CO2 in a humidified incubator.
10. Separation of Stellate Cells (0.5 hr)
- Unlabeled HSC pass the separation column during the MACS procedure. Collect the HSC fraction (see step 9.5, Figure 2.5). Perform cell counting as described in step 4.6.
- Seed HSC with a density of 5 x 104 cells/cm2 in cell culture vessels coated with rat tail collagen (see step 6.1) in Stellate Cell/Endothelial Cell culture medium (Table 2) and cultivate them at 37 °C, 5% CO2 in a humidified incubator.
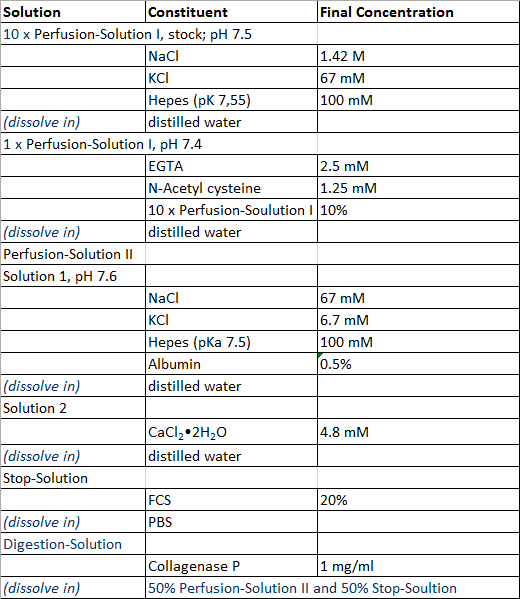
Table 1: Perfusion and isolation solution.
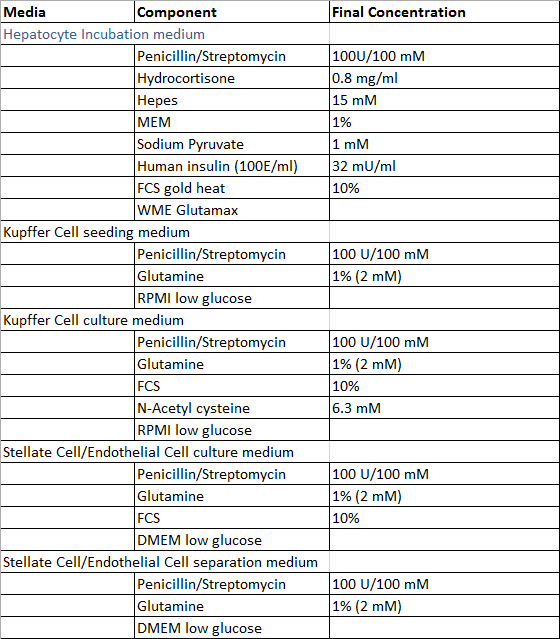
Table 2: Culture and isolation media.
The separation into a parenchymal and non-parenchymal fraction, using density gradient centrifugation as a clean-up procedure combined with the use of adherence properties and MACS leads to successful PHH and NPC isolation. PHH and NPC can be isolated in high quality and quantity. Figure 1 shows the representative setup of the equipment for liver perfusion and digestion. 10% FCS was added to the collagenase P containing Perfusion – Solution II to reduce proteolytic activity of proteases and to stabilize collagenase P activity in return. In consequence longer digestion times required for NPC isolation can be applied without a negative impact on viability of PHH.
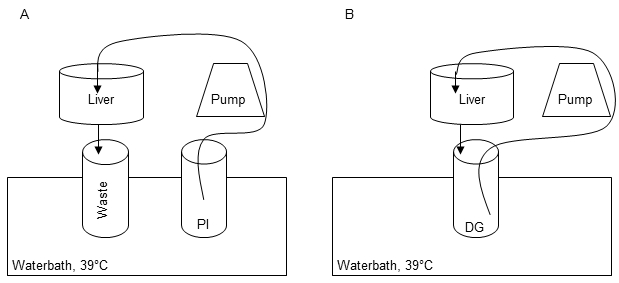
Figure 1: Perfusion and digestion setup. The first perfusion step is carried out in order to remove residual blood, warm up the tissue and remove Ca2+ to dissolve cell-cell-connections by using 1x Perfusions Solution I (PI) (A). Recirculation of Digestion-Solution (DG) is performed for digestion of the liver tissue during perfusion step II (B).
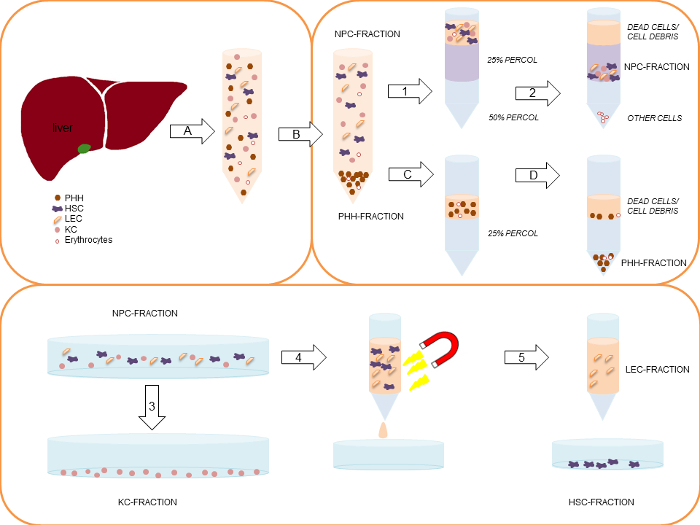
Figure 2: Simplified schematic representation of the complete PHH and NPC isolation process. Modified from Pfeiffer et al.1, 2014 with permission of Experimental Biology and Medicine. First, the liver tissue sample is perfused and digested by a two-step EGTA/collagenase P perfusion technique (A). The gained cell suspension is centrifuged initially at 50 x g, 5 min, 4 °C (B), to separate the larger PHH-fraction (pellet) from the smaller NPC-fraction (supernatant). In case of a PHH viability below 70%, the viable PHH fraction can be enriched by a density gradient centrifugation at 1,250 x g, 20 min, 4 °C (C) resulting in settling of the PHH at the bottom of the tube, while dead cells/ cell debris are located on top of the density gradient layer (D). Collected supernatants of the initial centrifugation (1) are centrifuged using two steps: 1) 300 x g, 5 min, 4 °C and 2) 650 x g, 7 min, 4 °C. After the first centrifugation KC are partly located in the supernatant. In this context the second isolation step is necessary. The gained cell pellets are pooled and re-suspended in HBSS. Subsequently, the cell suspension are carefully layered on top of a two-layer (25%/50%) density gradient. The layered density gradient tubes are centrifuged at 1,800 x g, 20 min, 4 °C (2). Dead cells on top of the 25% density gradient layer are discarded. NPC located between the interphase of the 25% and the 50% density gradient layer are collected and pooled. The NPC fraction is seeded on uncoated cell culture plastics. Using a 20 min incubation time (adherence separation step) KC are separated from other liver cell populations (3). LEC and HSC are separated by using the MACS-kit. Therefore the collected remaining liver cells in the supernatant are centrifuged at 300 x g, 5 min, 4 °C, and are labeled with CD31-conjugated MicroBeads (4). Only CD31-negative HSC pass the MACS separation column (5). CD31-positive LEC stick to the column. Finally the column is removed from the magnetic device and the CD31-positive LEC are eluted out of the column (5). Please click here to view a larger version of this figure.
The isolated PHH showed a yield of 14.2 x 106 ± 6.6 x 106 viable PHH/g liver tissue and a viability about 76.6 ± 4.2% (Table 31). Microscopically visible features were a typical large cytoplasmic volume in combination with lipid droplets and between one and four nuclei, (Figure 3A). The cell size varies between 20 to 30 µm in suspension.
KC were the most common cell type in the NPC fraction. We isolated approximately 1.9 x 106 ± 0.2 x 106 viable KC/g liver tissue with a viability of 92.8 ± 3.5% (Table 31). KC are very small cells (about 5 µm) with a low cytoplasm/nucleus ratio and typical microvilli on the surface (Figure 3B).
Finally we used the MACS separation technique to separate CD31-positive LEC from the remaining CD31-negative HSC. The yield of LEC was approximately 2.7 x 105 ± 0.1 x 105 viable LEC/g liver tissue and the achieved viability was 95.6 ± 2.8% (Table 31). Identification criteria are the multiple granulae and a size of about 10 µm in cell suspension (Figure 3C) as well as the characteristic spindle shape after a short cultivation time (Figure 3G).
The isolation process resulted in a HSC yield about 4.7 x 105 ± 0.2 x 105 viable HSC/g liver (N = 8) with a viability of 89.6 ± 3.8% (Table 31). Microscopically identifiable characteristics were a size of about 20 µm and a typical granulated appearance with a varying amount of lipid droplets (Figure 3D).

Table 3: Yields, viability and purity of isolated PHH and NPC. Three different donors were evaluated. Data are given as mean ± SD. This Table was published before in Pfeiffer et al.1, 2014 and is reprinted with permission from Experimental Biology and Medicine.
For the identification and determination of cell culture purity, every isolated cell fraction was treated with antibodies against cell type specific antigens. The cells were treated with fluorescent secondary antibodies and investigated by immunofluorescence microscopy. The purity was determined by counting positive fluorescent stained cells in relation to the total cell number visualized by Hoechst staining.
After 24 hr of cultivation PHH showed a characteristic polygonal shape and often polyploidy (Figure 3E). PHH were positive for CK 18 (Figure 3I) and showed a purity of 92.3 ± 3.2% (Table 31).
KC adhered within 20 min on cell culture plastic surfaces. After an incubation time of 24 hr small round cells with a prominent round cell nucleus were observed (Figure 3F). The surface protein CD68 was used to identify KC (Figure 3J). The purity of CD68 positive cells amounted to 81.0 ± 5.4% (Table 31).
Despite the MACS separation using CD31 labelling during the NPC separation it was still possible to stain isolated LEC with CD31. Therefore the isolated and cultivated LEC were stained with CD31 for identification and determination of purity. Additionally LEC showed immunoreactivity for the mesenchymal cell marker vimentin (Figure 3K). We observed approximately 81.0 ± 1.7% of positive stained cells (Table 31).
HSC with their typical prominent lipid droplets (Figure 3H) were marked by immunofluorescence staining for GFAP (Figure 3L). HSC purity was 93.0 ± 1.7% (Table 31).
Every cell fraction was counterstained with other NPC markers. All cell fractions contained a small number of other liver specific cell types, but were negative for the hepatocyte marker CK18 and the cholangiocyte marker CK19.
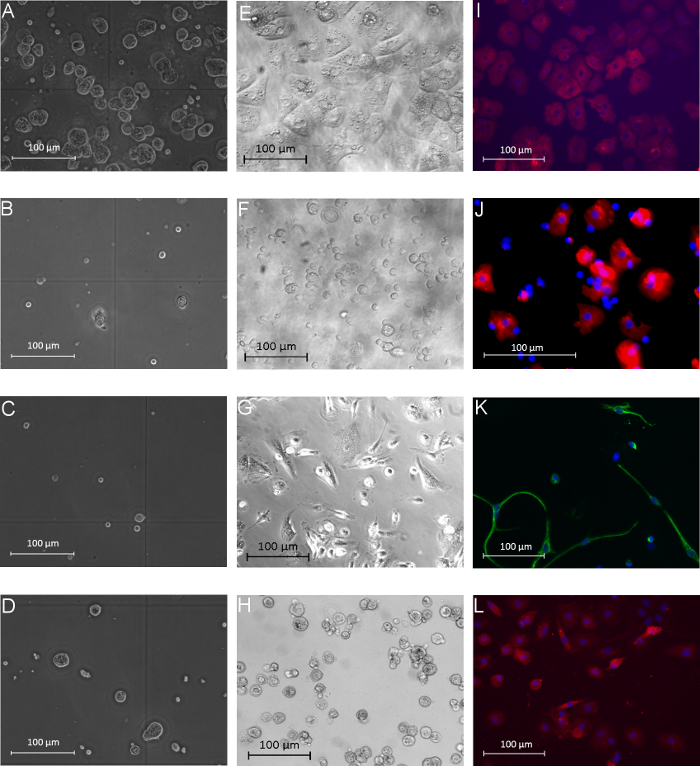
Figure 3: Morphology of human parenchymal and non-parenchymal liver cells in suspension and after adherence. The left column (A–D) shows the different isolated liver cell populations directly after the isolation process in phase contrast microscopy view: PHH (A), KC (B), LEC (C), and HSC (D). The middle column (E–H) presents images of isolated and cultivated PHH (E), KC (F), LEC (G) and HSC (H) after 24 hr of cultivation (phase contrast microscopy). Immunofluorescence-based characterization of the different cell fractions is shown in the last column: PHH showed positive signals for the hepatocyte marker CK18 (I, 24 hr after isolation), KC were positive for the marker CD68 (J, 24 hr after isolation), LEC showed positive signals for vimentin (K, 72 hr after isolation) and HSC were positive for GFAP (L, 72 hr after isolation). Cell nuclei were stained with Hoechst; magnification: 400X. Modified from Pfeiffer et al.1, 2014 with permission of Experimental Biology and Medicine. Please click here to view a larger version of this figure.
