Separation of lymphohematopoietic progenitors by FACS cell sorting
We here demonstrate results from sorting the necessary cell populations for downstream STR analysis. Bone marrow cells were stained with V450-conjugated anti-CD45, FITC-conjugated anti-CD36 and APC-conjugated anti-CD34. The population of interest is the megakaryocyte erythroid progenitors (MEP), nucleated cells responsible for the development of erythrocytes. These cells express CD36, but are negative for the leukocyte common antigen CD45 (Figure 1C). If necessary, these cells can be further differentiated based on their CD36 expression level. Several additional populations can be sorted to serve as controls. We sorted CD45+CD34+ lymphoid/myeloid precursors as another progenitor cell population and CD45+ cells with high SSC signal as mature myeloid cells (Figure 1D). Sorting was performed with a BD FACSAria I instrument. After sorting, erythroid progenitor cell purity was >85% (Figure 1E) and myeloid cell purity was >95% (Figure 1F).
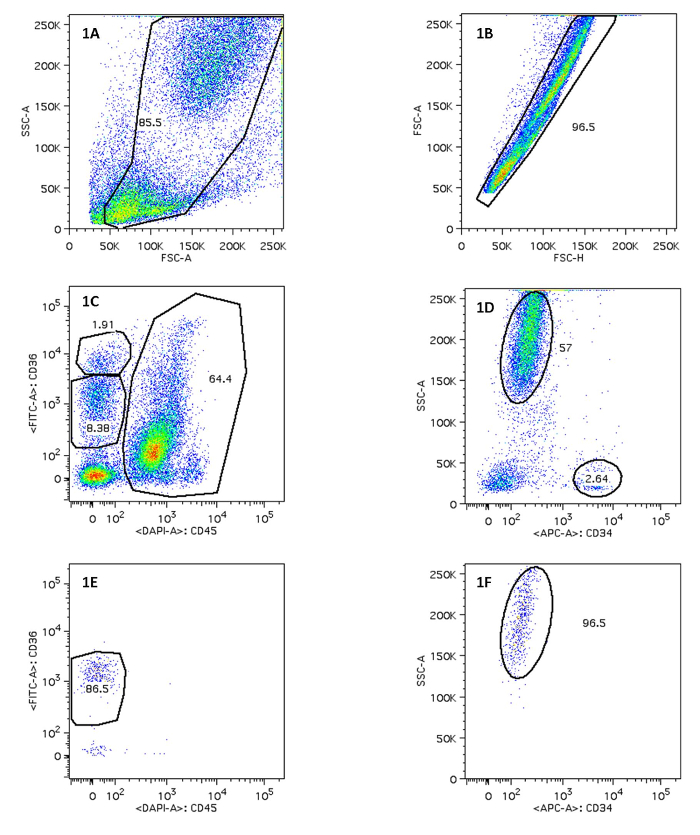
Figure 1. Sorting of Erythroid Progenitor Cells and Myeloid Progenitor Cells After FACS. Figure 1A/1B displays on FSC-A vs. SSC-A, FSC-H vs. FSC-A and the use of a polygon gate tool to select the population of interest. 1C indicates MEP on CD45 vs. CD36; 1D displays mature myeloid cells. The percentage of positive cells in 1E and 1F is shown. Please click here to view a larger version of this figure.
Generation of burst-forming units – erythroid colonies
Some cells derived from bone marrow and separated by CD34-specific magnetic beads appear to proliferate and differentiate. After a 14-day incubation period on the semi-solid medium colonies are formed. Stimulation with various concentrations of EPO greatly augmented the formation of prominent red (erythroid) colonies (Figure 2).
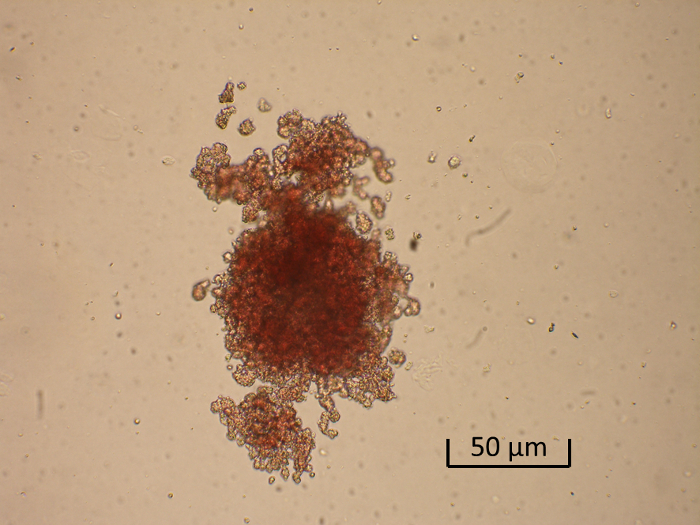
Figure 2. Generation of a Burst-forming Unit-erythroid (BFU-E). Shown is a representative low-power photomicrograph of a BFU-E colony. Please click here to view a larger version of this figure.
| MNC: | |||
| 1X EPO | 2X EPO | 4X EPO | |
| CFU-E | 2 | 2 | 2 |
| BFU-E | 9 | 10 | 8 |
| CFU-GM | 30 | 30 | 29 |
| CFU-GEMM | 1 | 1 | 2 |
| CD34+ cells: | |||
| 1X EPO | 2X EPO | 4X EPO | |
| CFU-E | 1 | 1 | 0 |
| BFU-E | 5 | 11 | 6 |
| CFU-GM | 25 | 26 | 32 |
| CFU-GEMM | 0 | 2 | 3 |
Table 1. Analysis of Colonies in Unsorted Bone Marrow Cells and CD34+ Selected Cells. Colonies are scored according to their morphology with an inverted microscope at 40X magnification in a culture dish marked with a scoring grid. For the purposes of our assay, the colonies are classified in four categories: colony-forming unit-erythroid (CFU-E), burst-forming unit-erythroid (BFU-E), granulocyte-macrophage progenitor cells (CFU-GM) and multipotential progenitor cells (CFU-GEMM).
Analysis of chimerism
Chimerism analysis is performed using the AmpFlSTR Profiler Plus Kit (Applied Biosystems, California) according to the manufacturer's protocol on ABI Prism 310 Genetic Analyzer (Applied Biosystems, California). Analysis is performed with GeneMapper Software (Applied Biosystems, California). Loci D21S11, D7S820, FGA and vWA were used to calculate results (percentage of recipient- and donor-specific DNA).
| Sample | Recipient DNA | Donor DNA |
| CD45 | 25% | 75% |
| CD36hi | 25% | 75% |
| CD36lo | 55% | 45% |
| CD34 | 25% | 75% |
Table 2. Percent Recipient and Donor DNA in Cells Obtained from Fluorescence-activated Cell Sorting of Bone Marrow Cells. Chimerism of donor cells in specific cellular lineages of the lymphohematopoietic compartment is given.
| Sample | Recipient DNA | Donor DNA |
| BM-MNC | 24.71% | 75.29% |
| BM-CD34 | 22.62% | 77.38% |
| BFU-E (BM-MNC) | 80.99% | 19.01% |
| CFU-GM (BM-MNC) | 0.00% | 100.00% |
| CFU-GEMM (BM-MNC) | 5.73% | 94.27% |
| BFU-E (BM CD34) | 30.13% | 69.87% |
| CFU-GM (BM CD34) | 0.00% | 100.00% |
| CFU-GEMM (BM CD34) | 9.87% | 90.13% |
Table 3. Percent Recipient and Donor DNA in Cells Obtained from the Clonogenic Assay. Chimerism of donor cells in specific cellular lineages of the lymphohematopoietic compartment is given.
