Reconstitution of Cell-cycle Oscillations in Microemulsions of Cell-free Xenopus Egg Extracts
Summary
We present a method for the generation of in vitro self-sustained mitotic oscillations at the single-cell level by encapsulating egg extracts of Xenopus laevis in water-in-oil microemulsions.
Abstract
Real-time measurement of oscillations at the single-cell level is important to uncover the mechanisms of biological clocks. Although bulk extracts prepared from Xenopus laevis eggs have been powerful in dissecting biochemical networks underlying the cell-cycle progression, their ensemble average measurement typically leads to a damped oscillation, despite each individual oscillator being sustained. This is due to the difficulty of perfect synchronization among individual oscillators in noisy biological systems. To retrieve the single-cell dynamics of the oscillator, we developed a droplet-based artificial cell system that can reconstitute mitotic cycles in cell-like compartments encapsulating cycling cytoplasmic extracts of Xenopus laevis eggs. These simple cytoplasmic-only cells exhibit sustained oscillations for over 30 cycles. To build more complicated cells with nuclei, we added demembranated sperm chromatin to trigger nuclei self-assembly in the system. We observed a periodic progression of chromosome condensation/decondensation and nuclei envelop breakdown/reformation, like in real cells. This indicates that the mitotic oscillator functions faithfully to drive multiple downstream mitotic events. We simultaneously tracked the dynamics of the mitotic oscillator and downstream processes in individual droplets using multi-channel time-lapse fluorescence microscopy. The artificial cell-cycle system provides a high-throughput framework for quantitative manipulation and analysis of mitotic oscillations with single-cell resolution, which likely provides important insights into the regulatory machinery and functions of the clock.
Introduction
Cytoplasmic extracts prepared from Xenopus laevis eggs represent one of the most predominant models for the biochemical study of cell cycles, given the large volume of oocytes, the rapid cell cycle progression, and the capability of reconstituting mitotic events in vitro1,2. This system has allowed the initial discovery and mechanistic characterization of essential cell-cycle regulators like maturation-promoting factor (MPF) as well as downstream mitotic processes including spindle assembly and chromosome segregation1,2,3,4,5,6,7,8,9,10,11. The Xenopus egg extracts have also been used for detailed dissection of the regulatory networks of the cell cycle clock8,12,13,14 and for studies of the DNA damage/replication checkpoint15 and the mitotic spindle assembly checkpoint16,17,18.
These studies of cell cycles using the Xenopus egg extracts have mainly been based on bulk measurements. However, conventional bulk reaction assays may not mimic real cell behaviors, given a major discrepancy in their dimensions and subcellular spatial compartmentalization of reaction molecules. Moreover, bulk measurements of mitotic activities are prone to giving a limited number of cycles before quickly damping8. These disadvantages of bulk reactions have prevented the extract system to provide further understanding of complex clock dynamic properties and functions. Recent studies have encapsulated cell-free cytostatic factor-arrested (CSF) Xenopus extracts19,20 into size-defined cell-like compartments, which have helped elucidate how spindle size is modulated by the cytoplasmic volume. However, this in vitro system is arrested at metaphase of meiosis II by the action of cytostatic factor1, and a system capable of long-term sustained oscillations at the single-cell level is needed for further investigation of the cell cycle oscillator.
To study cell cycle oscillations with single-cell resolution, we have developed a cell-scale, high-throughput system for reconstitution and simultaneous measurement of multiple self-sustained mitotic oscillatory processes in individual microemulsion droplets. In this detailed video protocol, we demonstrate the creation of the artificial mitotic oscillation system by encapsulating cycling Xenopus laevis egg cytoplasm in microemulsions of sizes ranging from 10 to 300 µm. In this system, mitotic oscillations including chromosome condensation and de-condensation, nuclear envelope breakdown and reformation, and the degradation and synthesis of anaphase substrates (e.g., securin-mCherry in this protocol) were successfully reconstituted.
Protocol
All methods described here have been approved by the Institutional Animal Care and Use Committee (IACUC) of University of Michigan.
1. Preparation of Materials for Cell Cycle Reconstitution and Detection
- Gibson Assembly cloning for the plasmid DNA construction and mRNA purification of securin-mCherry
- Prepare three DNA fragments including a pMTB2 vector backbone, securin, and mCherry through polymerase chain reaction (PCR) and gel purification21,22.
- Measure the concentrations of fragments using a fluorospectrometer. Combine 100 ng of backbones with securin and mCherry inserts at a 1:1:1 molecular ratio and add deionized water to adjust the total volume of reaction to 5 µL.
- Prepare 6 mL of 5x isothermal assembly reaction buffer with 3 mL of 1 M Tris-HCl (pH 7.5), 300 µL of 1 M MgCl2, 600 µL of 10 mM dNTP, 300 µL of 1 M DTT, 1.5 g of PEG-8000, and 20 mg of NAD23.
- Add the combined fragments to a 15 µL of 1x isothermal assembly reaction buffer aliquot. Incubate the reaction in a PCR reaction tube at 50 °C for 15 minutes.
- Make a 0.8% agarose gel and run with a voltage of 10 V/cm to verify the annealing efficiency of these fragments.
- Purify the constructed plasmid and elute using 15 µL of RNase-free water. Transform 1 µL of the purified plasmid DNA into 50 µL of DH5α competent E. coli cells24 for future use.
- Extract and purify the plasmid DNA of securin-mCherry from the DH5α competent E. coli cells.
- Transcribe the linearized securin-mCherry plasmid DNA into mRNA and purify the mRNA. Use a spectrophotometer to verify that the concentration of mRNA is more than 100 ng/µL and the ratio of absorbance at 260 nm and 280 nm is about 2.0.
- Preparation of demembranated Xenopus laevis sperm DNA
NOTE: Adapted from Murray 19911.- Euthanize adult male Xenopus laevis25 and dissect testes from four male frogs26,27.
- Place testes from four frogs in the same 100 mL Petri dish. Rinse the testes three times in 50 mL of cold 1x MMR solution (0.1 M NaCl, 2 mM KCl, 1 mM of MgCl2, 2 mM CaCl2, 5 mM HEPES, 0.1 mM EDTA).
- Remove excess blood and fat from the testes in a 100 mL petri dish and then wash twice in cold nuclear preparation buffer (NPB) (250 mM sucrose, 15 mM HEPES, pH 7.4, 1 mM EDTA, pH 8.0, 0.5 mM spermidine trihydrochloride, 0.2 mM spermine tetrahydrochloride).
- Remove the nuclear preparation buffer (NPB) and cut testes into pieces with lengths smaller than 0.2 cm using sharp dissecting scissors in a 100 mL Petri dish. Add 8 mL of cold NPB to the testes for further breakdown.
- Use nylon mesh fabrics with a pore size of 70 µm to filter the testes and collect sperm sample into a 15 mL conical tube. Spin down the collected sperms at 1,730 x g for 10 minutes at 4 °C and remove supernatant.
- Supply the sperm cells with 1 mL of NPB and 50 µL of 10 mg/mL lysolecithin and incubate at room temperature for 5 min to demembranate sperm cells.
- Wash the demembranated sperm DNA with 10 mL of cold NPB with 3% BSA solution three times.
- Measure the density of sperm DNA with a hemocytometer.
- Freeze the sperm DNA in 5 µL aliquots in liquid nitrogen and store at -80 °C. The optimal concentration of sperm DNA is approximately 105 nuclei/µL.
- Protein purification of GFP-Nuclear Localization Signal (GFP-NLS)
- Transform the GST-tagged GFP-NLS plasmid into BL21 (DE3) competent E. coli cells5.
- Incubate the cells without antibiotics at 37 °C for 1 h and then spread on a LB agarose plate with 100 µg/mL ampicillin.
- Incubate the plate at 37 °C for 14 to 18 hours to grow the cells into single colonies. Pick and inoculate single colonies into 4 mL of LB media with 100 µg/mL ampicillin. Shake the cells at 37 °C for 12 hours.
- Prepare the autoclaved YT media (16 g of Bacto-tryptone, 10 g of Bacto-yeast extract, 5 g of NaCl) and heat the YT media to 37 °C.
- Inoculate 1 mL of the overnight incubated cells in LB media into 250 mL of preheated YT media with 100 µg/mL ampicillin and shake for 2 hours to increase cell density.
- Measure cell optical density (OD) every 30 minutes, using a spectrophotometer at a wavelength of 600 nm. Add 0.1 mM IPTG into cells to induce GFP-NLS protein expression once the OD value reaches 0.1.
- Shake the cells at 18 °C overnight to reduce cell growth rate and allow better protein expression and synthesis.
- Spin down the cells at 34,524 x g at 4 °C for 5 minutes and remove supernatant.
- Resuspend the cell pellets in 40 mL of PBS buffer (supplied with 800 µL of 50 mg/mL lysozyme, 100 µL of 0.2 M PMSF, 13.2 µL of a 10 mg/mL mix of leupeptin, pepstatin, and chymostatin, and 8 µL of 0.5 M EDTA) and incubate at 4 °C for 20 minutes.
- Break down cells using a sonicator at a frequency of 20 kHz for 4 x 30 s, with 30 s intervals between each sonication, to release expressed GFP-NLS protein.
- Spin at 20,000 x g for 35 minutes to remove the broken cells.
- Use affinity chromatography to extract and purify GFP-NLS protein.
- Wash the 1.5 mL bead slurry three times with 15 mL of PBS buffer and incubate 250 mL of lysate with beads for 4 hours at 4 °C with inversion.
- Spin down the beads at 2000 x g for 5 min and add 10 mL of wash solution (25 mM Tris base, pH 7.5, 500 mM NaCl, and 5 mM DTT) to beads. Incubate beads in 700 µL of elution buffer (50 mM Tris-HCl, pH 8.8, 20 mM reduced glutathione, and 5 mM DTT) with rotation at 4 °C. Collect an aliquot of the elution with a volume of 50 µL.
- Measure the concentration of collected protein by running a Coomassie blue gel28.
- Elute the proteins using PD-10 columns with 3 mL of storage buffer (20 mM HEPES, 150 mM KCl, 10% glycerol, pH 7.7) through buffer exchange.
2. Preparation of Cycling Xenopus Extracts
NOTE: The overall procedure of preparing the extracts is illustrated in Figure 1A. Adapted from Murray 19911.
- Inject three mature female Xenopus laevis with 66 IU pregnant mare serum gonadotropin (PMSG) 10 days before laying eggs.
- Inject these Xenopus frogs with 500 IU of human chorionic gonadotropin (HCG) to induce egg laying 18 hours before the preparation of cycling extracts.
- Discard laid eggs overnight. Based on experiments (data not shown), extracts prepared from the freshly squeezed eggs generate longer-lasting activities than extracts prepared from laid eggs from the same female frog. Squeeze the eggs of each female frog into 100 mm Petri dishes containing 10 mL of 0.2x MMR buffer and inspect under a stereo microscope.
- Discard the batches of eggs that have unclear boundaries between animal and vegetal poles or have irregular white spots on top of animal poles.
- Choose the batch of eggs from one female frog presenting a homogeneous population of eggs with clearly differentiated animal and vegetal poles and transfer the eggs into a 600 mL beaker.
- Pour out excess 0.2x MMR buffer and gently add 250 mL of 2 g per 100 mL cysteine (pH 7.7) in 1x extract buffer (100 mM KCl, 0.1 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, 50 mM sucrose, pH 7.7) to eggs.
- Shake the eggs vigorously by hand and remove the jelly coats of eggs over three washes with a total volume of 800 mL of cysteine buffer for 3 minutes or until eggs aggregate and settle at the bottom of the beaker, indicating that jelly coat removal is successful.
- Wash eggs with 1 L of 0.2x MMR solution over four times.
- Pick and discard eggs that turn white using a glass transfer pipette.
- Pour out the MMR buffer and supply eggs with 200 mL of 0.1 µg/mL calcium ionophore A23187 solution in 0.2x MMR buffer for activation.
- Use the wide opening side of a glass pipette to stir the eggs gently until the eggs are activated and remove calcium ionophore solution.
- Check the activation efficiency after eggs settle down at the bottom of the beaker. Well-activated eggs have approximately 25% of the animal pole and 75% of the vegetal pole.
- Wash the activated eggs twice with 50 mL of 1x extract buffer supplemented with protease inhibitors (10 µg/mL each of leupeptin, pepstatin, and chymostatin).
- Carefully transfer eggs to a 0.4 mL snap-cap microtube and then pack eggs through centrifugation at 200 x g for 60 seconds, and then 600 x g for 30 seconds.
- Remove extra buffer on the top of eggs using a glass Pasteur pipette to reduce the dilution of extracts.
- Crush eggs at 15,000 x g for 10 minutes at 4 °C.
- Cut the tubes with a razor blade to separate three layers of crushed eggs and keep the crude cytoplasm in the middle layer.
- Transfer the crude cytoplasm into new ultracentrifuge tubes, and supplement with protease inhibitors and cytochalasin B at 10 µg/mL each.
- Centrifuge the crude cytoplasm again at 15,000 x g at 4° C for 5 minutes to further purify cytoplasm by removing excess lipid and yolk.
- Keep freshly prepared extracts on ice.
3. Droplet Generation
- 2D glass chamber preparation
NOTE: The rectangle glass tubes serve as chambers that confine the droplets in a single 2-dimensional plane, allowing for easier observation and data collection.- Cut rectangle glass tubes with an inner dimension of 2 mm x 100 µm into 4 mm long pieces. Use the sharp edge of a whetstone, mark the desired boundaries on the outer surface of the glass tube with light etchings, then gently apply pressure on both sides of the etchings to break off pieces of glass tubes.
- Pre-heat a dry bath incubator to 95 °C. Take out the heating block and place it into a polycarbonate vacuum desiccator.
- Place a 1.5 mL microcentrifuge tube containing 30 µL of trichloro(1H,1H,2H,2H-perfluorooctyl)silane in the heating block. Lay the cut glass tubes inside the vacuum desiccator at approximately 5 cm to the heating block.
- Connect the desiccator to a vacuum pump. Turn on the pump for 1 minute to reach the desired vacuum level, then close the 3-way valve to seal the desiccator and let the trichloro(1H,1H,2H,2H-perfluorooctyl)silane vapor coat the inner wall of the glass tubes. This will create a hydrophobic environment for stabilizing the droplets.
- Leave the glass tubes inside the desiccator for overnight coating. The tubes will be ready for use the next day.
- Droplet generation and imaging
NOTE: The procedures of generating droplets and imaging are illustrated in Figures 1B and 1C.- Supply extracts prepared in step 2 with 10 ng/µL securin-mCherry mRNA for the simple cytoplasmic-only cell cycle experiments. Alternatively, supply extracts with demembranated sperm chromatin (250 per µL of extract), 10 µM GFP-NLS protein, and 10 ng/µL securin-mCherry mRNA to create droplets exhibiting periodic nuclei morphology changes.
- In a 1.5 mL microcentrifuge tube, mix 20 µL of extract supplied with recombinant protein or mRNA with 200 µL of 2% perfluoropolyether-polyethylene glycol (PFPE-PEG) fluorosurfactant in HFE7500 fluorinated oil.
- Generate droplets using a vortex mixer. The range of droplet sizes can be modulated by vortex speed and duration. To create droplets with radii ranging from 50 to 200 µm, set the vortex speed at 56 x g (3,200 rpm with a radius of 4.9 mm) and gently press the microcentrifuge tube containing the extract and surfactant mixture on the mixer for 3.5 seconds. Visually inspect if the droplets are uniform in size.
- Use a 20 µL pipette to transfer the droplets floating on top and an equal volume of PFPE-PEG surfactant into a PCR tube. Dip the glass tube prepared from step into the droplet layer in 1.5 mL microcentrifuge tube and wait until droplets have filled approximately 75% of the tube, then push the tube down into the surfactant layer until the tube is completely filled. This will lower the droplet density and allow the droplets to form one single layer inside the chamber without overlapping or shape distortion caused by overcrowding.
- Fill a glass bottom dish with a diameter of 40 mm with mineral oil. Immerse the droplet-filled tubes in oil by gently pushing them down using a fine tweezer.
- After loading all samples, use a glass pipette to remove any visible debris or leaked droplets. Add more mineral oil if the tubes are not or will not be completely immersed in the imaging process.
- Conduct imaging of droplets on an epifluorescence microscope equipped with a motorized x-y stage (see Table of Materials). Use a 4X air objective for all imaging. For fluorescence imaging, use a 130 W mercury vapor short arc lamp as fluorescence light source for illumination, and a GFP filter set (excitation 482/35 nm and emission 514/LP nm) and an mCherry filter set (excitation 562/40 nm and emission 641/75 nm) for imaging GFP-NLS and securin-mCherry signals.
- Record time-lapse videos in the bright field and multiple fluorescence channels every 6 to 9 minutes until the droplets lose their activity.
4. Image Processing and Data Analysis
- Segmentation and tracking of droplets
- Import recorded data in the format of ".tif" or ".oif" into image analysis software (see Table of Materials).
- Segment single droplets using the bright field channel. The bright field image will show droplets as bright circles with dark boundaries. Apply thresholding to the image. Add a tracking surface to each droplet by tuning the threshold between dark and bright pixels so that each surface covers the whole area of the corresponding droplet.
- Automatically track the segments between adjacent frames using the autoregressive motion algorithm. Tune the acceptable maximum traveling distance of a droplet between two adjacent frames to be smaller than the radius of most droplets to accurately track small droplets.
- Visually check all tracks across the entire video to detect incorrect segmentations that segment the empty space between droplets instead of actual droplets. Manually delete incorrect tracks.
- Segment and track background area without any droplets to obtain the background fluorescence intensity. To improve signal to noise, subtract background intensity from the fluorescence intensity of droplets at each time frame.
- Export measured parameters for each track over time, including the mean and standard deviation of the fluorescence intensity and the droplet area into ".csv" files.
- Data analysis of droplet size and oscillation period
- Load the ".csv" files into R to create plots of fluorescence intensity over time for each droplet. Record droplet ID into a ".csv" file.
- Import the ".csv" files exported from the analysis software and the ".csv" file with a list of droplet ID into Matlab and save as ".mat" files for further data analysis.
- Calculate the volume of a compressed droplet based on the exported droplet area information after segmentation and the height of glass chamber19. Use the volume to calculate the radius of droplet as a sphere.
- Extract the time points of peaks and troughs using peak/trough detection algorithm. Perform manual corrections on the position of peaks and troughs to ensure they are accurately detected.
- To calculate the cell-cycle period, calculate the interval time between two adjacent peaks. Take the average of all intervals for each droplet as its cell-cycle period.
Representative Results
In Figure 2, we show that this protocol produces mitotic oscillations in both simple, nuclear-free cells as well as complicated cells with nuclei, where the oscillator drives the cyclic alternation of nuclei formation and deformation. The nuclei-free droplets generate mitotic oscillations up to 30 undamped cycles over the time span of 92 hours, as indicated by the periodic synthesis and degradation of a fluorescence reporter securin-mCherry (Figure 2A). Securin is a substrate of the anaphase promoting complex APC/C. The degradation of securin-mCherry during anaphase thus indicates the activity of APC/C.
To examine the ability of the mitotic oscillator to drive downstream mitotic events, we additionally supplied demembranated sperm chromatin, purified green fluorescent protein-nuclear localization signal (GFP-NLS), and Hoechst 33342 dyes to the cytoplasmic extracts. Figure 2B shows the autonomous alternation of distinct cell-cycle phases, interphase (blue bars) and mitosis (red bars), during which the changes of nuclear morphology are driven by the self-sustained mitotic oscillator. All mitotic events, i.e., the chromosome decondensation and condensation reported by Hoechst 33342 (Figure 2B, first row), the nuclear formation and nuclear envelope breakdown by GFP-NLS (Figure 2B, second row), and the activity of the cell-cycle oscillator by the synthesis and degradation of securin-mCherry (Figure 2B, third row), coincided with each other. Images were collected using time-lapse multi-channel fluorescence microscope. Our experimental results demonstrate that the reconstructed cell-cycle system is capable of reconstructing a mitotic oscillator of Cdk1 and APC/C, which can drive a series of downstream events including nuclear envelope breakdown and reformation, and chromosome morphology change.
Figure 3 shows the oscillations indicated by the mean fluorescence intensity of securin-mCherry before and after noise removal. Peaks and troughs became more pronounced after removing the background noise, especially for the first two oscillations, indicating that noise removal can help improve signal to noise for further analysis.
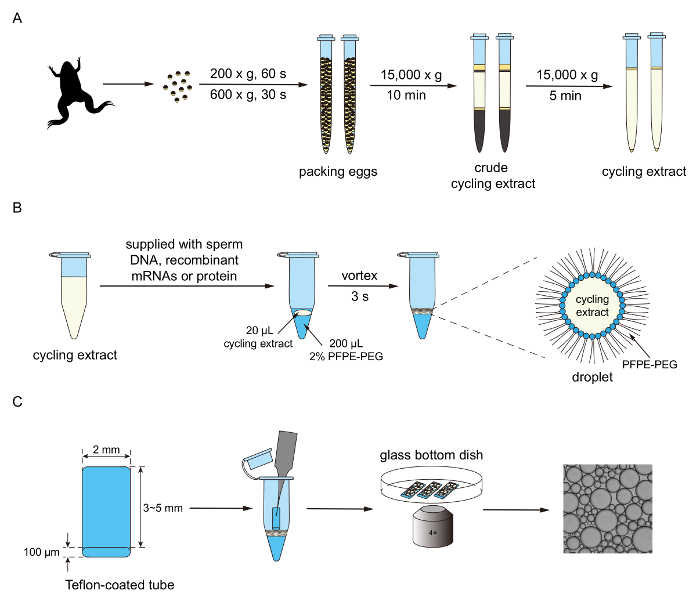
Figure 1. Experimental procedure for generating droplet-based mitotic cells. A. Cytoplasmic extracts are collected through ultra-centrifugation. B. The extracts are supplied with multiple components for the purpose of reconstituting and reporting mitotic oscillations. Using a vortexing method, the extracts and surfactant oil are mixed to generate droplets. C. Droplets are loaded into a trichloro(1H,1H,2H,2H-perfluorooctyl)silane-coated tube and then imaged through an epi-fluorescence microscope. Please click here to view a larger version of this figure.
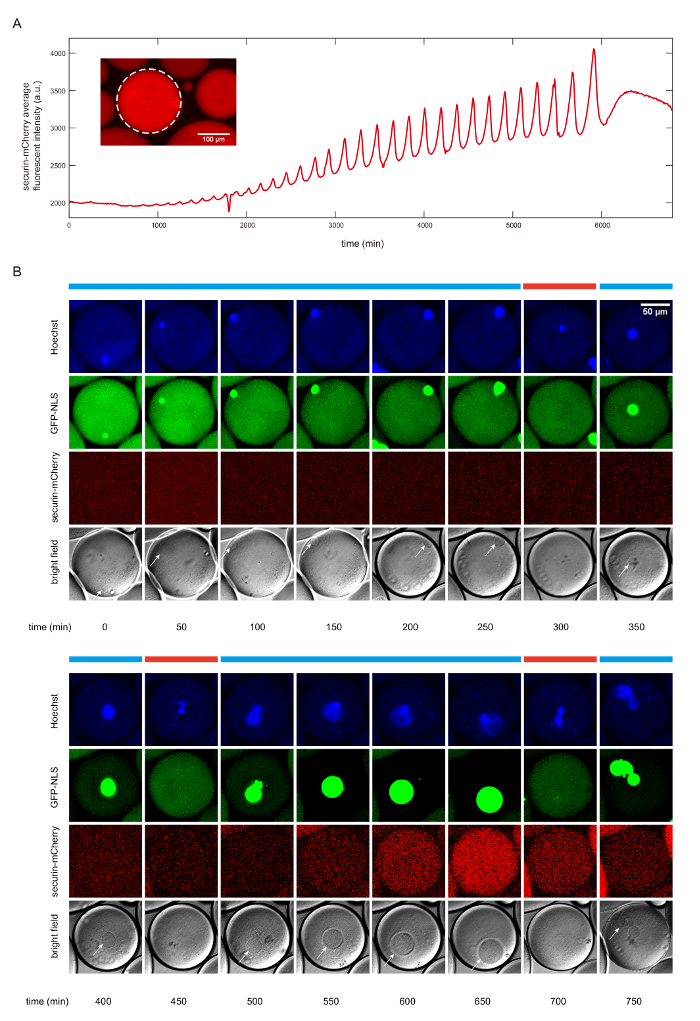
Figure 2. The detection of cell cycle dynamics using fluorescent reporters. A. The time course of the mean securin-mCherry fluorescence intensity of the selected droplet, indicating 30 undamped oscillations over a course of 92 hours. The insert figure shows the fluorescence image with the selected droplet framed by white dotted circle. The scale bar is 100 µm. B. Time series of snapshots of a droplet in fluorescence channels (top three rows) and bright field (the last row). The cyclic progression of the cell cycle clock and its downstream mitotic processes are simultaneously tracked by multiple fluorescent reporters. The anaphase substrate securin-mCherry indicates activity of the clock regulator APC/C, the Hoescht stain indicates chromosomal morphology changes, and the GFP-NLS indicates nuclear envelope breakdowns and reformations. Nuclear envelopes (highlighted by white arrows) are also detectable in the bright field images, matching the localization of GFP-NLS indicated nuclei. The scale bar is 50 µm. Interphase and mitotic phase are respectively denoted by the blue bars and red bars above the images. Please click here to view a larger version of this figure.
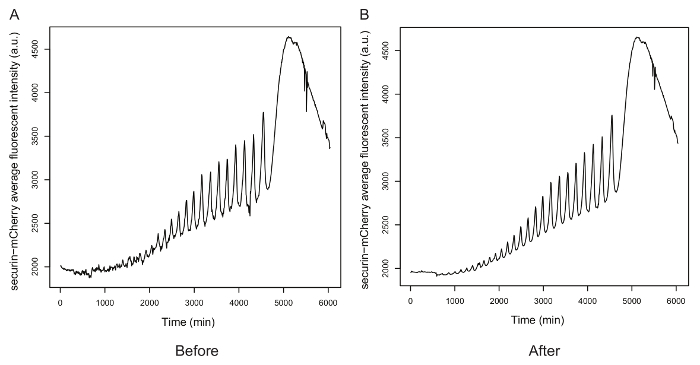
Figure 3. Background subtraction for the mean fluorescence intensity of securin-mCherry. A. The oscillation time course before background noise removal. B. The oscillation time course after noise removal. Please click here to view a larger version of this figure.
Discussion
We have presented a novel method for developing a high-throughput artificial cell system that enables in vitro reconstitution and long-term tracking of self-sustained cell-cycle oscillations at the single-cell level. There are several critical steps that make this method successful. First, freshly squeezed Xenopus eggs with a good quality, compared with laid eggs, tend to produce extracts with longer-lasting oscillation activity. Second, encapsulation of extracts within the surfactant-stabilized microenvironments is crucial to preserve the oscillation activity. Third, incubation of the oil droplets into a thin tube confines the droplets within a single layer, making it easier for image processing and long-term tracking. Fourth, coating the tube inner wall with trichloro(1H,1H,2H,2H-perfluorooctyl)silane as an anti-adhesive to lower the surface energy helps preserve the extract activity. Fifth, sealing the tubes with oil would prevent droplet flow. The oil-sealing method can also prevent evaporation and further preserve the extract activity. These together improve the success rate of in vitro reconstruction of robust oscillations in droplets and the accuracy of segmentation and tracking of these droplets in a long-term.
To generate microemulsion droplets, we have employed both a simple vortexing method, similar to a few other studies29,30,31, as well as the more commonly used microfluidics techniques19,20 (data not shown). Both methods were able to successfully generate microemulsions in a high-throughput manner. One limitation associated with the vortexing method is that the droplet size is not uniformly controlled. For studies that require a well-defined size with minimized size variation, microfluidics techniques should be used for better control of droplet sizes. Here, we have chosen the vortexing method over the microfluidic method for two reasons. First, it has simple and easy-to-follow procedures, which would allow labs without access to microfluidic techniques or nanofabrication facilities to adapt this method easily. Second, the vortexing method is more efficient and economic for investigating size-dependent behaviors of the oscillator. It can generate a wide distribution of droplet sizes with radii varying from several micrometers up to 300 micrometers, while the microfluidic technique can only generate a narrow distribution centered on a predefined size. The range of droplet sizes generated with this method matches the wide range of cell sizes resulting from the characteristic reductive cell divisions in the cleavage stage of early embryos such as Xenopus and zebrafish.
The present method has significantly improved the sustainability of oscillations and the throughput of analyzing complex clock dynamics, compared with existing methods of reconstituting biological oscillators in well-mixed bulk solution8,32, which tend to produce quickly damped oscillations. We have shown that this method has significantly improved the lifespan of oscillations from a few cycles in bulk reaction8 to over 30 cycles in the microenvironment of droplets (Figure 2A). Additionally, bulk reactions lack the similarity to the actual cell dimensions and cannot recapitulate the spatial organization achieved by functional compartmentalization in real cells. Having overcome these limitations of bulk reactions, our artificial cell system provides an essential platform for investigating challenging questions such as the cellular heterogeneity and the stochasticity of oscillators, which are otherwise impossible to explore.
Moreover, this method provides flexibility in the degree of complexity to which cells can be built. To avoid any interference from the complicated nuclear dynamics, we can reconstitute a minimal, cytoplasmic-only cell-cycle system that contains no nuclei. This simple and cytoplasmic-only oscillator exhibits self-sustained oscillations significantly better than some existing synthetic oscillators. By supplying sperm chromatin, we can also reconstitute more complex cells with nuclei that alternate between self-assembly at interphase and nuclear deformation at the mitotic phase in a rhythmic manner. The activity of the mitotic oscillator as well as the downstream nuclear events can be simultaneous measured by multi-channel fluorescence imaging and single-cell tracking.
The method also enables a high degree of manipulation in droplet size and cell-cycle speed. The vortexing technique allows for generation of droplets with radii in a wide range, which will benefit the study of cell-size dependent behaviors of cell cycles. In addition, the cell-cycle speed can be modulated by supplying different concentrations of cyclin B1 mRNAs33, which would allow us to study the tunability function of the cell-cycle clock.
With these advantages, the in vitro reconstructed cell-cycle platform, enabling visualization, manipulation, and analysis of single-cell dynamics, will likely pave the way for new insights into the more complicated stochastic behaviors of the cell cycle clock. The method may also be generalizable to the in vitro studies of other oscillators that traditionally rely on bulk reactions.
Divulgazioni
The authors have nothing to disclose.
Acknowledgements
We thank Madeleine Lu for constructing securin-mCherry plasmid, Lap Man Lee, Kenneth Ho and Allen P Liu for discussions about droplet generation, Jeremy B. Chang and James E. Ferrell Jr for providing GFP-NLS construct. This work was supported by the National Science Foundation (Early CAREER Grant #1553031), the National Institutes of Health (MIRA #GM119688), and a Sloan Research Fellowship.
Materials
| Xenopus laevis frogs | Xenopus-I Inc. | ||
| QIAprep spin miniprep kit | QIAGEN | 27104 | |
| QIAquick PCR Purification Kit (250) | QIAGEN | 28106 | |
| mMESSAGE mMACHINE SP6 Transcription Kit | Ambion | AM1340 | |
| BL21 (DE3)-T-1 competent cell | Sigma-Aldrich | B2935 | |
| Calcium ionophore | Sigma-Aldrich | A23187 | |
| Hoechst 33342 | Sigma-Aldrich | B2261 | Toxic |
| Trichloro | Sigma-Aldrich | 448931 | Toxic |
| (1H,1H,2H,2H-perfluorooctyl) silane | |||
| PFPE-PEG surfactant | Ran Biotechnologies | 008-FluoroSurfactant-2wtH-50G | |
| GE Healthcare Glutathione Sepharose 4B beads | Sigma-Aldrich | GE17-0756-01 | |
| PD-10 column | Sigma-Aldrich | GE17-0851-01 | |
| VitroCom miniature hollow glass tubing | VitroCom | 5012 | |
| Olympus SZ61 Stereo Microscope | Olympus | ||
| Olympus IX83 microscope | Olympus | ||
| Olympus FV1200 confocal microscope | Olympus | ||
| NanoDrop spectrophotometer | Thermofisher | ND-2000 | |
| 0.4 mL Snap-Cap Microtubes | E&K Scientific | 485050-B | |
| PureLink RNA Mini Kit | ThermoFisher(Ambion) | 12183018A | |
| Fisherbrand Analog Vortex Mixer | Fisher Scientific | 2215365 | |
| Imaris | Bitplane | Version 7.3 | Image analysis software |
Riferimenti
- Murray, A. W. Cell cycle extracts. Methods in Cell Biology. 36, 581-605 (1991).
- Hannak, E., Heald, R. Investigating mitotic spindle assembly and function in vitro using Xenopus laevis egg extracts. Nature Protocols. 1, 2305-2314 (2006).
- Murray, A. W., Solomon, M. J., Kirschner, M. W. The role of cyclin synthesis and degradation in the control of maturation promoting factor activity. Nature. 339, 280-286 (1989).
- Yang, Q., Ferrell, J. E. The Cdk1-APC/C cell cycle oscillator circuit functions as a time-delayed, ultrasensitive switch. Nature Cell Biology. 15, 519-525 (2013).
- Chang, J. B., Ferrell, J. E. Mitotic trigger waves and the spatial coordination of the Xenopus cell cycle. Nature. 500, 603-607 (2013).
- Trunnell, N. B., Poon, A. C., Kim, S. Y., Ferrell, J. E. Ultrasensitivity in the Regulation of Cdc25C by Cdk1. Molecular Cell. 41, 263-274 (2011).
- Kim, S. Y., Ferrell, J. E. Substrate competition as a source of ultrasensitivity in the inactivation of Wee1. Cell. 128, 1133-1145 (2007).
- Pomerening, J. R., Kim, S. Y., Ferrell, J. E. Systems-level dissection of the cell-cycle oscillator: bypassing positive feedback produces damped oscillations. Cell. 122, 565-578 (2005).
- Pomerening, J. R., Sontag, E. D., Ferrell, J. E. Building a cell cycle oscillator: hysteresis and bistability in the activation of Cdc2. Nature Cell Biology. 5, 346-351 (2003).
- Telley, I. A., Gaspar, I., Ephrussi, A., Surrey, T. Aster migration determines the length scale of nuclear separation in the Drosophila syncytial embryo. The Journal of Cell Biology. 197, 887-895 (2012).
- Telley, I. A., Gaspar, I., Ephrussi, A., Surrey, T. A single Drosophila embryo extract for the study of mitosis ex vivo. Nature Protocols. 8, 310-324 (2013).
- Tsai, T. Y. C., Theriot, J. A., Ferrell, J. E. Changes in Oscillatory Dynamics in the Cell Cycle of Early Xenopus laevis Embryos. PLoS Biology. 12, e1001788 (2014).
- Chang, J. B., Ferrell, J. E. Mitotic trigger waves and the spatial coordination of the Xenopus cell cycle. Nature. 500, 603-607 (2013).
- Yang, Q., Ferrell, J. E. The Cdk1-APC/C cell cycle oscillator circuit functions as a time-delayed, ultrasensitive switch. Nature Cell Biology. 15, 519-525 (2013).
- Kumagai, A., Dunphy, W. G. Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in Xenopus egg extracts. Molecular Cell. 6, 839-849 (2000).
- Chen, R. H., Murray, A. Characterization of spindle assembly checkpoint in Xenopus egg extracts. Methods in Enzymology. 283, 572-584 (1997).
- Chen, R. H., Waters, J. C., Salmon, E. D., Murray, A. W. Association of spindle assembly checkpoint component XMAD2 with unattached kinetochores. Science. 274, 242-246 (1996).
- Minshull, J., Sun, H., Tonks, N. K., Murray, A. W. A MAP kinase-dependent spindle assembly checkpoint in Xenopus egg extracts. Cell. 79, 475-486 (1994).
- Good, M. C., Vahey, M. D., Skandarajah, A., Fletcher, D. A., Heald, R. Cytoplasmic Volume Modulates Spindle Size During Embryogenesis. Science. 342, 856-860 (2013).
- Hazel, J., et al. Changes in cytoplasmic volume are sufficient to drive spindle scaling. Science. 342, 853-856 (2013).
- Garibyan, L., Avashia, N. Research Techniques Made Simple: Polymerase Chain Reaction (PCR). The Journal of Investigative Dermatology. 133, e6 (2013).
- Hecker, K. H., Roux, K. H. High and low annealing temperatures increase both specificity and yield in touchdown and stepdown PCR. BioTechniques. 20, 478-485 (1996).
- Gibson, D. G., et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature Methods. 6, 343-345 (2009).
- Froger, A., Hall, J. E. Transformation of Plasmid DNA into E. coli Using the Heat Shock Method. Journal of Visualized Experiments. 6, e253 (2007).
- Torreilles, S. L., McClure, D. E., Green, S. L. Evaluation and refinement of euthanasia methods for Xenopus laevis. Journal of the American Association for Laboratory Animal Science. 48, 512-516 (2009).
- Sive, H. L., Grainger, R. M., Harland, R. M. Isolating Xenopus laevis Testes. Cold Spring Harbor Protocols. 2007, (2007).
- Showell, C., Conlon, F. L. Egg Collection and In vitro Fertilization of the Western Clawed Frog Xenopus tropicalis. Cold Spring Harbor Protocols. 2009, (2009).
- Wilson, C. M. . Methods in Enzymology. 91, 236-247 (1983).
- Schutze, T., et al. A streamlined protocol for emulsion polymerase chain reaction and subsequent purification. Analytical Biochemistry. 410, 155-157 (2011).
- Weitz, M., et al. Diversity in the dynamical behaviour of a compartmentalized programmable biochemical oscillator. Nature Chemistry. 6, 295-302 (2014).
- Ho, K. K., Lee, J. W., Durand, G., Majumder, S., Liu, A. P. Protein aggregation with poly(vinyl) alcohol surfactant reduces double emulsion-encapsulated mammalian cell-free expression. PloS One. 12, e0174689 (2017).
- Nakajima, M., et al. Reconstitution of circadian oscillation of cyanobacterial KaiC phosphorylation in vitro. Science. 308, 414-415 (2005).
- Guan, Y., et al. A robust and tunable mitotic oscillator in artificial cells. eLife. 7, (2018).
