Two sets of data as control experiments are shown in Figure 221. Using 2,2,2-trifluoroethanol as reference compound, logP values were obtained for 2-fluoroethanol and 3,3,3,2,2-pentafluoropropanol as -0.75 and +1.20, respectively (Figure 2A). Subsequently, the lipophilicity of 2-fluoroethanol was determined again but with 3,3,3,2,2-pentafluoropropanol as the reference (using its previous experimentally measured logP value +1.20). The measured logP value was -0.76, which only had a difference of 0.01 logP units when compared with the value measured using 2,2,2-trifluoroethanol as reference.
Likewise, for cis-2,3-difluoro-1,4-butanediol, the difference in measured logP values by using 2-fluoroethanol and its trans isomer is also very small (0.01 logP units, Figure 2B). This demonstrated that the selection of reference compound does not have impact on the logP measurement. In addition, a rather small standard deviation (<0.01) indicated good reproducibility of our method.
Using our method, a series of compounds with known logP values was measured as shown in Table 1. The difference between literature data and the values measured using our method is shown in the last column of the Table. Overall, the experimentally obtained logP values (at 25 °C) have good to excellent accordance with the literature values, which further validated our method.
Additional selected examples21 were shown in Figure 3. All these non-UV-active aliphatic compounds (from fluorinated carbohydrates to fluorohydrins) can be easily measured with our method.
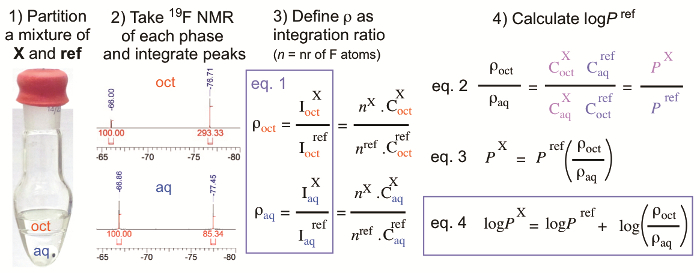
Figure 1: Principle of the logP determination method. This figure has been reproduced with permission from Wiley-VCH Verlag GmbH & Co. KGaA.21. This shake-flask method is based on 19F NMR spectroscopy. A reference compound is used for partition experiment. Aliquots for both n-octanol and water phase were taken for NMR experiment. Integration ratios between reference compound and the compound to be measured are obtained for the determination of logP value. Detailed mathematical deduction of equations, which leads to the final equation for measurement, are also given. Please click here to view a larger version of this figure.
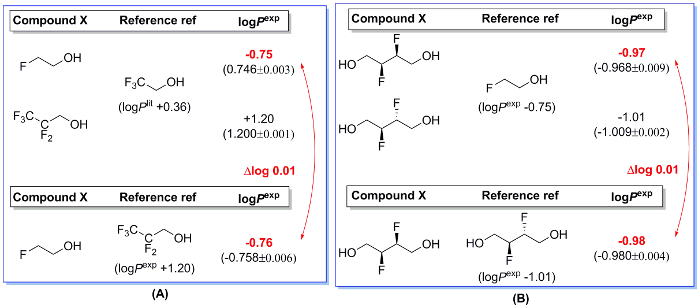
Figure 2: Examples for internal validation21. Two sets of control experiments, using two different reference compounds to measure logP value of one compound, were conducted. The logP difference between those experiments is negligible. Standard deviation (<0.01) from experiments run in triplicate shows good reproducibility of the method. Please click here to view a larger version of this figure.
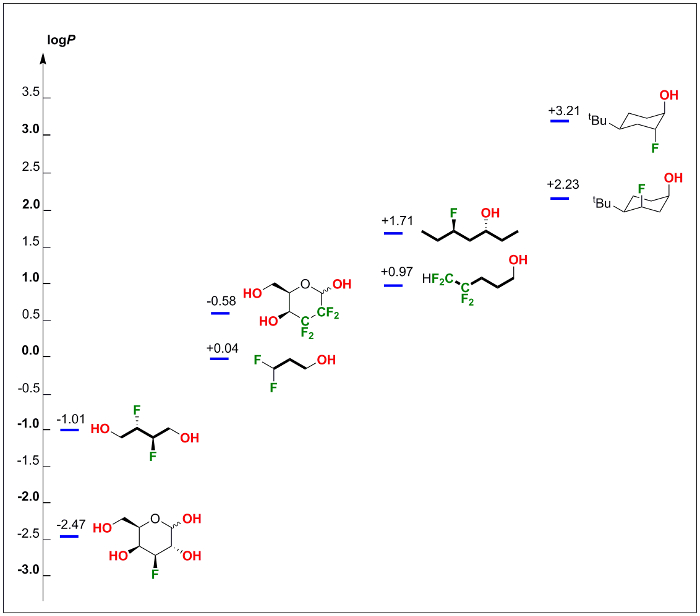
Figure 3: Further selected examples of logP measurement using our method. Applying this method, logP values for 8 fluorinated compounds (such as fluorinated carbohydrates, acyclic alkanols and conformationally restricted fluorohydrins) were obtained. Please click here to view a larger version of this figure.
Table 1: Comparison between literature data and the experimental logP values using our method21. logP values for 14 fluorinated compounds (with known logP data) were measured using this new method. The reference compounds used for each measurement were also tabulated. Comparision (logP) between literature values and logP results from our method demonstrated good accuracy of this method. a2,2,2-Trifluoroethanol (TFE), 2-Fluoroethanol (FE); bAveraged logP value from at least three experiments; cExperimentally measured logP value by our method (-0.75) was used as reference. Please click here to download this file.
