1. Protein and reagent preparation
NOTE: This protocol includes an example workflow for labeling a protein with DEPC. Some conditions and reagent concentrations listed may vary based on the protein of choice.
- Prepare all reagent solutions in 1.5 mL microcentrifuge tubes.
- Prepare a protein solution of desired concentration, usually in the range of tens of µM, in a 10 mM 3-(N-morpholino)propanesulfonic acid (MOPS) buffer at pH 7.4. Alternatively, prepare a buffer-exchange existing protein solution in 10 mM pH 7.4 MOPS if the sample contains a nucleophilic buffer that would be reactive with DEPC. Other buffers (e.g., phosphate buffered saline) can also be used, as long as they do not have nucleophilic functional groups.
- Prepare a 100 mM DEPC solution in dry acetonitrile (ACN) by pipetting 1.45 µL of the stock 6.9 M DEPC solution into 98.55 µL of the ACN.
NOTE: There is no one set of concentrations that will work with every protein, though the optimal concentrations can be estimated based on the number of His and Lys residues23. For example, with 50 µL of a 50 µM β2-microglobulin solution, react the protein with 0.2 µL of the 100 mM DEPC for a final DEPC concentration of 200 µM (equal to 4x the protein concentration) to ensure the desired molar ratio using this general example (Table 1). DEPC labeling is a 2nd order reaction, so changing either the concentration of protein or DEPC in the reaction mixture will change the labeling rate. - Prepare 1 M imidazole solution by weighing out 10 mg of imidazole and dissolving in 146.9 µL of HPLC-grade water.
2. Covalent labeling of intact protein
- Set water bath temperature to 37 °C and wait for the bath to reach a stable temperature.
NOTE: Reagent concentrations and volumes for an example labeling protocol can be found in Table 1. - In a new microcentrifuge tube, mix MOPS buffer and protein solution in volumes listed in Table 1.
- To the protein and buffer add 0.2 µL of the DEPC solution, making sure to properly mix the resulting solution, and then place the tube containing the reaction mixture into the 37 °C water bath for 1 minute.
NOTE: The volume of ACN added should not exceed 1% of the total reaction volume to avoid perturbation of the protein's structure during the labeling reaction. Reaction time is up to the user, although a 1 minute reaction under the example conditions minimizes overlabeling and the potential hydrolysis of DEPC14. - After 1 minute, remove the tube containing the reaction mixture from the water bath and quench the reaction with 1 µL of the 1 M imidazole solution to scavenge the remaining unreacted DEPC.
NOTE: The final concentration of imidazole in the reaction mixture should equal 50x the concentration of DEPC in the reaction mixture. This will ensure that remaining unreacted DEPC is scavenged.
3. Preparation of protein digest for bottom-up LC-MS
NOTE: Choose digestion conditions that are amenable to the protein of interest. Common steps involve unfolding the protein and reducing and alkylating any disulfide bonds.
- Unfold the protein by adding an appropriate unfolding reagent to the reaction mixture.
NOTE: Common unfolding agents include ACN, urea, and guanidine hydrochloride (GuHCl). - Prepare solutions of Tris(2-carboxyethyl)phosphine (TCEP) and iodoacetamide (IAM) by weighing out 5 mg of each and dissolving them in new microcentrifuge tubes in 174.4 and 270.3 µL of 10 mM pH 7.4 MOPS buffer, respectively, for the reduction and alkylation steps.
- Reduce disulfide bonds by adding 2 µL of the 100 mM TCEP (final concentration of 2 mM in reaction mixture) solution to the reaction mixture and reacting for 3 minutes at room temperature.
NOTE: The final concentration of TCEP should equal 40x the protein concentration per disulfide bond present in the solution. - Alkylate reduced cysteines with 4 µL of the 100 mM IAM solution (final concentration of 4 mM in reaction mixture) for 30 minutes in the dark. IAM is light-sensitive and will decompose under direct light.
NOTE: The final concentration of IAM in solution should be twice the concentration used for TCEP, or 80x the protein concentration per disulfide bond. - Digest the protein with an appropriate enzyme such as trypsin or chymotrypsin. A 10:1 protein:enzyme ratio for a 3-hour digestion at 37 °C with immobilized enzyme at a shaking rate of 300 strokes/min is typically sufficient for DEPC-labeled proteins. See Discussion.
- After digestion, separate the immobilized enzyme from the digested peptides by centrifugation at 12,000 rpm for 5 minutes.
- Analyze the sample immediately by LC-MS/MS or flash-freeze the sample with liquid nitrogen to minimize sample degradation and label loss. Store the flash-frozen samples at < -20 °C until ready for LC-MS/MS analysis.
4. LC-MS/MS Analysis
NOTE: Standard LC-MS/MS parameters for bottom-up proteomics can be used to identify labeled sites on the proteolytic peptide fragments. A general example is outlined below.
- Separate the DEPC-labeled peptides using a reversed-phase C18 stationary phase. Use a typical LC mobile phase of two solvents: (A) water + 0.1% formic acid and (B) ACN + 0.1% formic acid using a gradient (e.g., Figure 2) to achieve the best separation of peptides.
NOTE: The separation time can be optimized based on sample complexity, and mobile phase flow rate depends on whether capillary or nano LC is used. - Use a mass spectrometer capable of doing on-line LC-MS and MS/MS to identify DEPC modification sites on the peptide. In our experiments, we have successfully used several types of mass spectrometers. Any mass spectrometer capable of automatically performing MS/MS of many peptides during the course of an LC-MS analysis should be suitable. Relevant MS parameters include: ESI source voltage = -4000 V for regular ESI; -2000 V for nanospray; Orbitrap resolution = 60,000; Dynamic exclusion duration = 30 s; MS/MS activation type: CID, ETD, or both; Mass scan range = 200-2,000; Automatic Gain Control = 4.0E5 (MS1 in Orbitrap) and 5.0E4 (MS2 in linear quadrupole ion trap).
- Load and inject the digested, labeled protein sample into the LC system and start the LC-MS/MS acquisition. If the sample has been flash-frozen, thaw before analysis. Divert the LC effluent to waste for the first 5 minutes to avoid excessive salts from getting into the ESI source.
NOTE: A 5 µL injection loop is generally utilized, allowing for injection of approximately 2.5 µg of protein to the LC-MS/MS. This is dependent on the loading conditions of the LC as to not clog the sample injector.
5. Data analysis
- Identify DEPC label sites and quantify peptide peak areas using appropriate software for the mass spectrometer that is used.
- Include DEPC addition (72.02 Da) and carbamidomethylation (57.02 Da) as variable modifications. Additional search parameters for the MS/MS analysis are as follows: Maximum missed cleavages = 3; Fragment ion types = b and y; Precursor m/z tolerance = 10 ppm (this value should be higher if a quadrupole ion trap mass spectrometer is used); Fragment m/z tolerance = 0.5 Da (this value should be lower if a high-resolution mass spectrometer is used for a product ion scan); Precursor charge = 1-4.
NOTE: Different database search algorithms have different scoring systems, and many can have difficulty identifying DEPC-modified peptides because modification levels can be low. Adjusting the score cutoff may be necessary to identify more labeled peptides. If so, then manual interrogation of the MS/MS data should be used to verify low-scoring peptides. The product of the hydrolysis of the DEPC label is not included in the data searching because the hydrolyzed DEPC is no longer reactive toward nucleophilic side chains. - Determine residue-level modification percentages using the chromatographic peak areas of the modified and unmodified versions of the peptides.
NOTE: Any peptide containing the modified residue of interest must be considered and all charge states that are included must be present in all the measured samples. Peptides having different ionization efficiencies and eluting at different times causes this value to be a relative rather than absolute measure of the modification of a specific site.
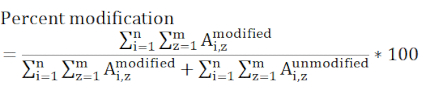
where Ai,z represents peak area of any given peptide (i) that contains the residue of interest and considers all detected charge states (z). - Determine if a labeling change between a control and experimental sample is significant using statistical evaluation. Three replicate measurements for each sample is typical, and t-tests are most commonly utilized with 95 or 99% confidence intervals.
Identifying DEPC modification sites and modification percentages
Mass addition due to covalent labeling can be measured at the (a) intact protein and (b) peptide levels8,9. At the intact level, a distribution of protein species with different numbers of labels can be obtained from direct analysis or LC-MS of labeled protein samples. To obtain higher resolution structural information (i.e., site-specific labeling data), measurements must be performed at the peptide level. After the labeling and quenching steps, the labeled proteins are subjected to bottom-up proteomic analysis (i.e., disulfide reduction, alkylation, proteolytic digestion, and LC-MS/MS). Figure 3 shows how the DEPC labeled sites are identified and their modification levels are calculated for a DEPC CL-MS experiment on the protein β-2-microglobulin (β2m)21. MS/MS is used to sequence the labeled peptides and pinpoint their DEPC labeled sites, while modification percentages are calculated from their relative peak areas in extracted ion chromatograms (See Figure 3A).
Protein surface mapping using DEPC CL-MS
Because of the relationship between protein topology and labeling rate, DEPC has been used to study changes in protein higher-order structure (HOS) and identify protein interaction sites8,9. An example is the effect of Cu(II) binding on the structure of β2m. DEPC modification rate coefficients of peptide fragments from unbound β2m and Cu(II)-bound β2m (b2m-Cu) can be measured (Table 2) by varying DEPC concentrations and generating second-order kinetic plots14,24. The DEPC reactivity of residues in β2m-Cu reveals significant labeling rate changes for His31, Ser33, and Thr4, while other sites, including different His residues, are statistically unchanged. These data are consistent with the fact that His31, but not other His residues in β2m, binds Cu causing structural changes near His31 (i.e., Ser33) and near the N-terminus (i.e., Thr4) (Figure 4)14,26,27.
DEPC CL-MS for identifying changes in HOS
DEPC labeling with MS detection is also a valuable tool for characterizing HOS changes to proteins, which has important implications for protein therapeutics, which are currently the fastest growing segment of the pharmaceutical market28. DEPC CL can identify specific protein regions that undergo structural changes upon thermal and oxidative stress18. After β2m is exposed to heat stress, many residues that undergo significant decreases in labeling extents (N-terminus, Ser28, His31, Ser33, Ser55, Ser57, Lys58) are clustered on one side of the protein, suggesting that this region of the protein undergoes a conformational change or possibly mediates aggregation (Figure 5A). In addition to these residues, after the protein is exposed to oxidative stress, other residues with a decrease in labeling (Ser11, His13, Lys19, Lys41, Lys94) form a cluster on another face of the protein, indicating that the oxidation-induced conformational changes occur elsewhere (Figure 5B). Other work from our group has also shown that DEPC CL-MS can detect and identify sites of conformational changes in heat-stressed monoclonal antibody therapeutics20.
DEPC CL-MS to study protein-protein interactions
Insight into protein-protein interaction sites and aggregation interfaces for amyloid-forming proteins can be obtained using DEPC labeling as well9. Decreases in the modification levels of residues upon oligomer formation can reveal the binding interfaces. DEPC CL-MS was used to characterize the pre-amyloid oligomers of β2m, which is the protein that forms amyloids in dialysis-related amyloidosis29, after initiating amyloid formation with Cu(II)15,16,26. Comparing the DEPC reactivity of the β2m monomer with the pre-amyloid β2m dimer that is formed 2 h after adding Cu(II) shows that nine residues undergo decreases in labeling while six residues undergo no change or slight increases in labeling (Figure 6A). Upon mapping these changes on the β2m, it is immediately apparent that the dimer interface involves the ABED β-sheets of β2m monomers (Figure 6B)15.
DEPC CL-MS to study protein-ligand binding
Ligand binding to proteins leads to decreases in the solvent accessibility of residues that interact with the ligand, and DEPC-based CL-MS can be used to identify ligand-binding sites on proteins. For example, the binding sites of epigallocatechin-3-gallate (EGCG) on β2m under amyloid-forming conditions can be identified by significant decreases in DEPC labeling at the residues buried by EGCG binding. In the presence of ECGC, Lys6 and Lys91 have lower DEPC modification percentages (Figure 7), and these residues are clustered in one region of the protein, indicating residue protection due to ligand binding. The N-terminus, Thr4, and His31, meanwhile, undergo increases in labeling extents (Figure 7), which are indicative of ECGC-induced structural changes and suggest that the Cu(II) binding sites on β2m (N-terminus and His31)14,30,31 may be disrupted as a result of ECGC binding32. In addition, the binding sites of the other two small-molecule inhibitors of β2m amyloid formation, rifamycin SV and doxycycline, have been identified using CL-MS19. In this study, however, results from DEPC labeling alone were not enough to map the binding sites with sufficient structural resolution. Results from three different CL reagents, namely DEPC, BD, and 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide – glycine ethyl ester (EDC/GEE) pair were necessary to better pinpoint the binding sites19.
Improving structural resolution and reducing label scrambling
Even though DEPC labeling causes the formation of a covalent bond, label loss due to hydrolysis can occur, especially for Ser, Thr, and Tyr residues (Figure 8). Label loss can be minimized by decreasing the time between DEPC CL reaction and LC-MS analysis using short proteolytic digestions (e.g., 2 h digestion with immobilized enzymes) rather than overnight digestions. Fast digestions result in more modified residues being measured, increasing the amount of protein structural information. For example, a 2 h digestion with immobilized chymotrypsin consistently leads to the identification of more DEPC-modified residues in β2m (Figure 9)17. With an overnight digestion about 75% of the Ser, Thr, Tyr, His, and Lys residues in β2m are detected, but when the time between labeling and LC-MS is reduced by using a 2 h digestion, 95% of these residues in β2m are detected. Most of the newly detected modified residues are Tyr and Thr residues that are prone to hydrolysis.
Over the course of our work with DEPC, we have noticed that in some proteins, Cys residues that from disulfide bonds are modified by DEPC, even though the disulfide bonds are intact during the DEPC labeling reactions. Such labeling of Cys residues occurs because free thiols can react with other modified residues in solution (Figure 10), leading to so-called "label scrambling" as the carbethoxy group is transferred to the Cys residue. Label scrambling decreases the modification levels at other residues and provides incorrect protein structural information. To avoid label scrambling, the free Cys thiols must be fully alkylated right after disulfide reduction33.
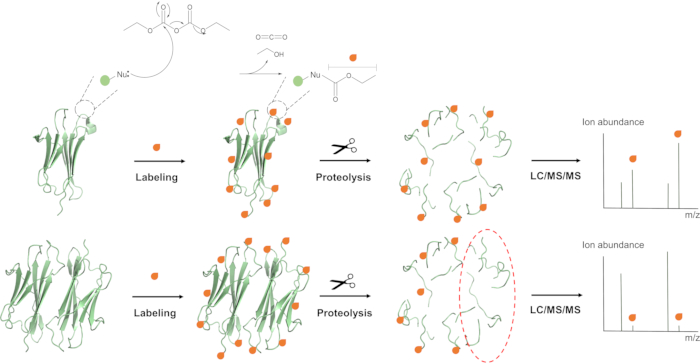
Figure 1: Covalent labeling-mass spectrometry (CL-MS). A monofunctional reagent is used to modify solvent accessible amino acids and can be used to provide site-specific information about conformational changes or interaction interfaces in proteins (top vs. bottom image). The modified protein is proteolytically digested, and the resulting peptides are analyzed by liquid chromatography (LC) in conjunction with MS and tandem MS (MS/MS). Figure has been adapted from Limpikirati, P., Liu, T., Vachet, R. W. Covalent Labeling-Mass Spectrometry for Studying Protein Structure and Interactions. Methods 144, 79-93 (2018). Please click here to view a larger version of this figure.
| Volume Used (μL) | Concentration in Solution (μM) | |
| Protein (100 μM, MOPS) | 50 | 50 |
| MOPS (10 mM, pH 7.4) | 48.8 | n/a |
| DEPC (100 mM) | 0.2 | 200 (=4x[protein]) |
| Imidazole (1 M) | 1 | 10,000 (=50x[DEPC]) |
| Total Volume | 100 |
Table 1. General DEPC labeling protocol.
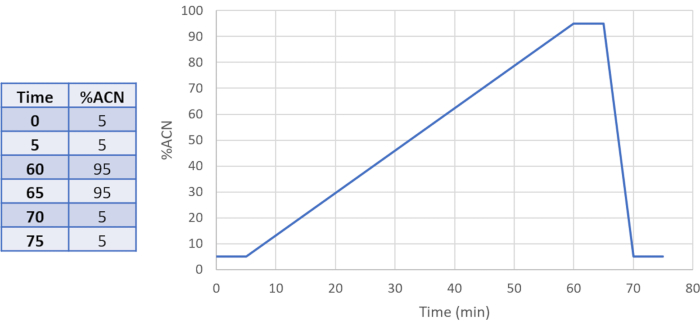
Figure 2. Example LC gradient for separation of peptides. Please click here to view a larger version of this figure.
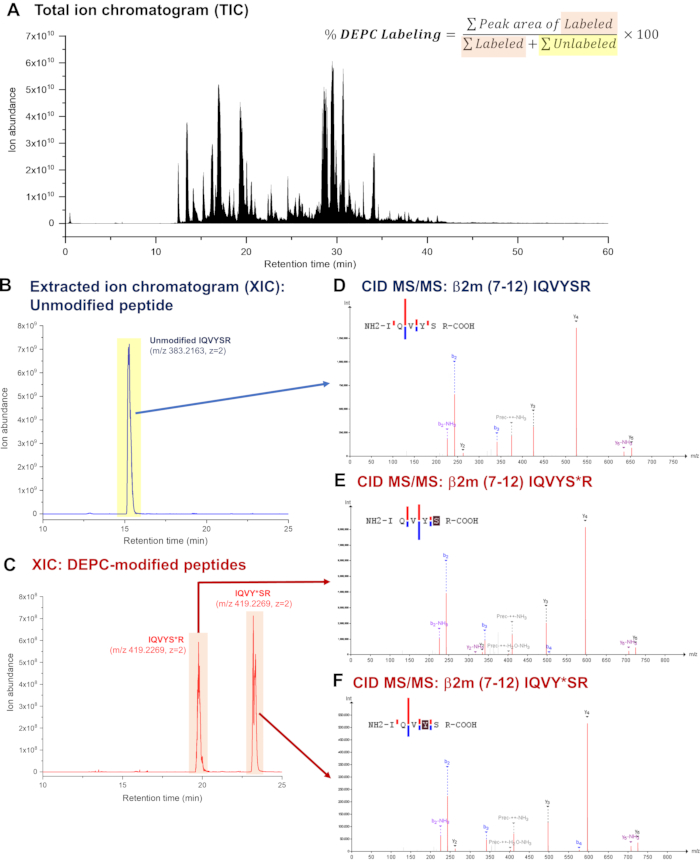
Figure 3 Illustration of how DEPC labeled sites are identified and their modification levels are calculated. After DEPC labeling and proteolytic digestion, (A) LC-MS analysis of the digested protein is performed. Peak areas of (B) unlabeled and (C) labeled peptides in a chromatogram are used to calculate the labeling percentage. During LC-MS, peptides are subjected to CID MS/MS. Tandem mass spectra of (D) unlabeled and (E) & (F) labeled peptides obtained at specific retention times are used for peptide sequencing and identification of DEPC labeled sites. Please click here to view a larger version of this figure.
| Residue | b2m | b2m-Cu |
| Thr4 | 0.082 ± 0.004 | 0.052 ± 0.009 |
| His13 | 0.041 ± 0.003 | 0.033 ± 0.005 |
| His31 | 0.010 ± 0.001 | 0.003 ± 0.001 |
| Ser33 | 0.010 ± 0.002 | 0.004 ± 0.001 |
| His51 | 0.036 ± 0.003 | 0.030 ± 0.004 |
| Ser88 | 0.029 ± 0.007 | 0.020 ± 0.006 |
Table 2 Site-specific DEPC modification rate coefficients (k, M-1s-1) for b2m in the absence and presence of Cu(II).
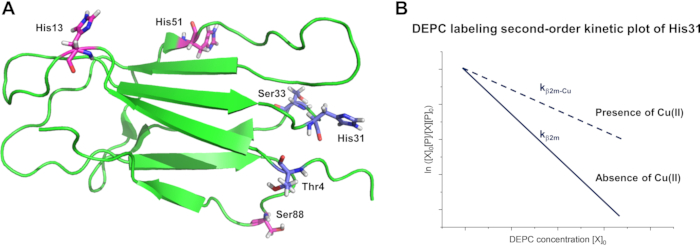
Figure 4 Using DEPC CL-MS to determine the effect of Cu(II) binding on the structure of β2m: (A) Protein surface mapping of the residues with significant decreases in DEPC labeling rates (blue), indicating changes in their solvent accessibility upon Cu(II) binding, and those with no significant changes in labeling rate (magenta) (PDB accession code 1JNJ). (B) Example site-specific second-order kinetic plots of the reaction of β2m with different concentrations of DEPC in the presence and absence of Cu(II). The labeling rate coefficient (k) can be obtained from the slope of the kinetic plot. Please click here to view a larger version of this figure.
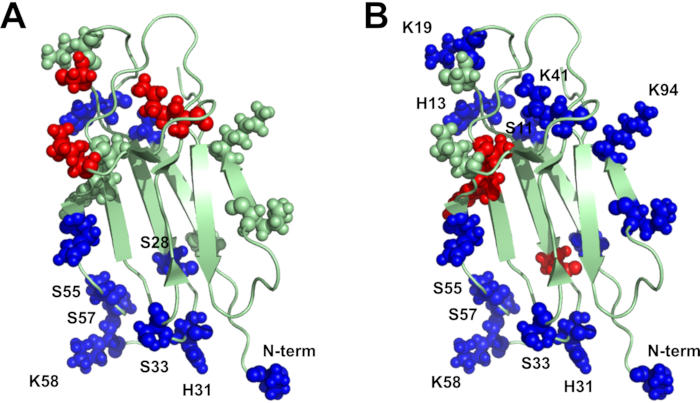
Figure 5 Covalent labeling results for stressed vs. native β2m: (A) Heat stress – heating at 75 °C for 24 h and (B) oxidative stress – with 3% H2O2 for 24 h. Significant changes in modification percentages are shown in blue (decrease in labeling) and red (increase in labeling) while residues with no significant labeling changes are shown in pale green (PDB accession code 1JNJ). Please click here to view a larger version of this figure.
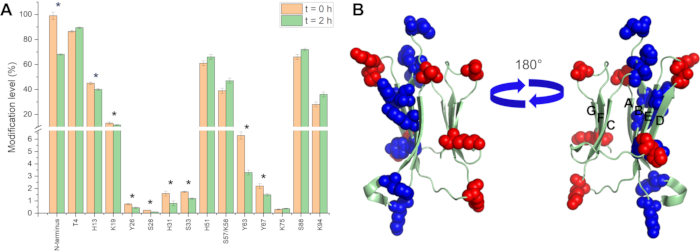
Figure 6 Using DEPC CL-MS to determine the dimer interface for the pre-amyloid dimer of β2m: (A) Summary of DEPC modification level changes for modified residues in the monomer (i.e., t = 0) and dimer 2 h after adding Cu(II). Residues with significant decreases in labeling extent are denoted by an asterisk (*). (B) Covalent labeling results mapped on the β2m structure. Residues with decreased labeling are shown in blue and residues with no changes or slight increases in labeling are shown in red (PDB accession code 1LDS). Please click here to view a larger version of this figure.
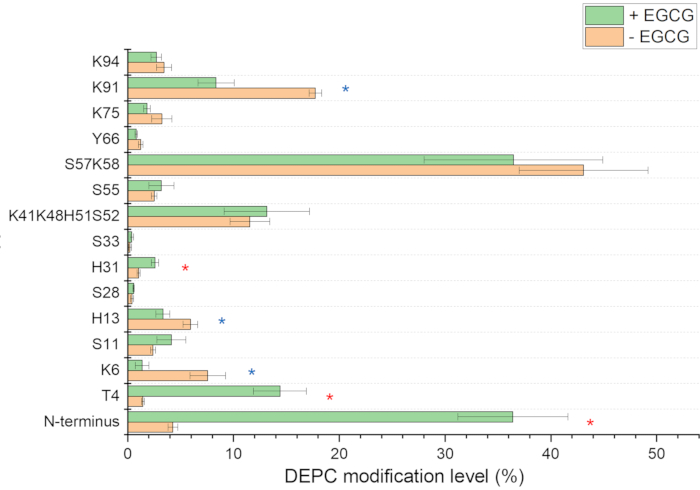
Figure 7 Covalent labeling results for EGCG-bound vs. unbound b2m. The DEPC modification percentages are shown for the displayed residues. Statistically significant differences in the covalent labeling percentage are denoted by an asterisk (*). Increases in labeling upon ECGC binding are shown in red while decreases are shown in blue. Please click here to view a larger version of this figure.
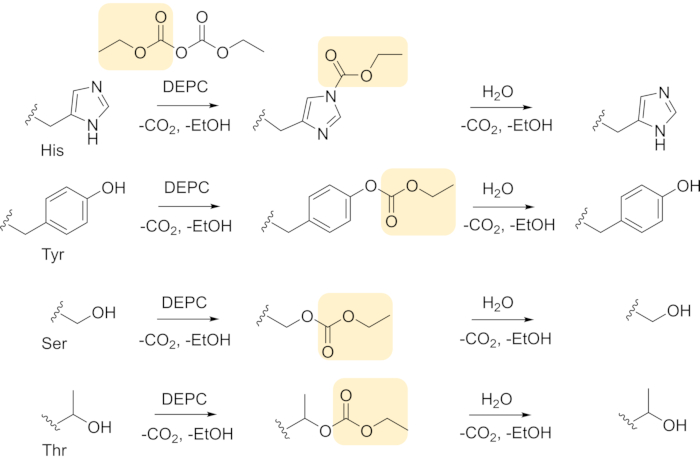
Figure 8 DEPC covalent labeling reactions (nucleophilic acyl substitution) and label loss (hydrolysis of carbethoxylated residues) Please click here to view a larger version of this figure.
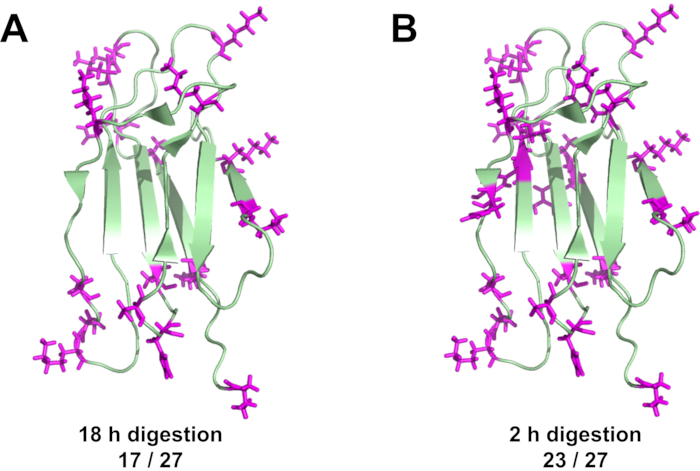
Figure 9 Mapping of the measured modification sites on β2m. (A) Labeled sites after conventional overnight digestion, and (B) labeled sites after a 2 h digestion with immobilized chymotrypsin. Please click here to view a larger version of this figure.
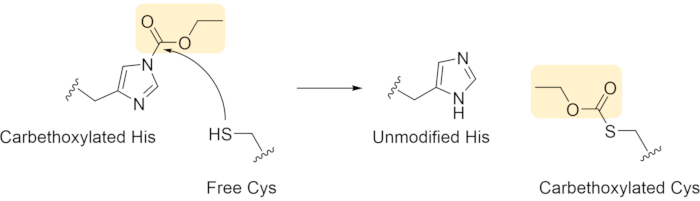
Figure 10 Hypothesized mechanism of DEPC label scrambling (Cys capture of carbethoxylated His) Please click here to view a larger version of this figure.
