Flow cells containing the actomyosin bundle systems are of a standard design, consisting of a microscope slide and an etched coverslip separated by a channel made from double-sided sticky tape (Figure 1). The assay is then built from the coverslip up using staged introductions as described in the protocol. The final assay consists of template rhodamine-labeled actin filaments; the desired myosin concentration (1 μM was used for the representative results in Figure 2 and Figure 3); biotinylated, Alexa Fluor 488-labeled actin filaments; 1 μm streptavidin beads; the oxygen scavenging system; ATP; and APB buffer. Multiple bundles will be formed per flow cell, and the actin concentrations described above give adequate spacing between bundles to ensure no unwanted interactions. This also facilitates obtaining multiple force measurements per flow cell to increase data acquisition efficiency. Force profiles should be reproducible within a flow cell and from flow cell to flow cell.
While the protocol above is geared toward the use of a commercial optical trapping setup, the flow cell and assay presented here could be easily utilized for a different commercial instrument or custom-built optical trapping setup coupled with a microscope or microscope stage and possessing fluorescence imaging capabilities. Once all flow cell additions are complete according to the above protocol, the actomyosin bundles on the slide (Figure 1) are ready for immediate measurement. The flow cell is added to the optical trap microscope stage, multiple bead calibration measurements are acquired, and bundles are identified through fluorescence colocalization of the bundle filaments. A bead bound to a bundle is trapped, and the displacement and corresponding force measurement begins. The user can observe the acquisition of data in real time on the computer monitor. Depending on the concentration of myosin used in the flow cell, the bundle could begin exhibiting substantial movement immediately, or it may take 30 s-1 min to effectively see an increase in displacement/force.
A representative force trace is shown in Figure 3A where the myosin motors exhibit a steady ramp in force followed by a plateau. It is typical to see these types of traces develop over 2-5 min. However, it is also possible to measure actomyosin bundles that do not generate any net force (Figure 3B). These traces appear as baseline noise or exhibit no substantial net increase in force over 90 s. This is likely due to a low local concentration of motor that does not permit productive sliding, or the bundle is in an unfavorable parallel orientation where the plus and minus ends of the filaments are aligned.
As the contents of the flow cell can be susceptible to degradation from the incident illumination and trapping laser, local heating on the slide over time, and generation of radical oxygen species, it is strongly advised to not use the same flow cell for more than 1 h. For maximum efficiency, it is suggested to have another assay incubating while acquiring data. Displacement/force trace can be exported from the optical trapping software into Excel, Matlab, Igor, or other data management programs for further filtering and analysis. Data that can be extracted from such optical trapping ensemble/bundle experiments include different types of force generation profiles (baseline, ramp/plateau) under varying assay conditions, velocity of force generation, maximum force generation, ensemble kinetic and stepping behavior through step sizes and dwell times between steps or teams of steps, as well as duty ratio. The user can also alter the assay conditions to compare how adding different types of myosin motors, adding actin binding proteins, or changing buffer conditions influence these ensemble force generation characteristics.
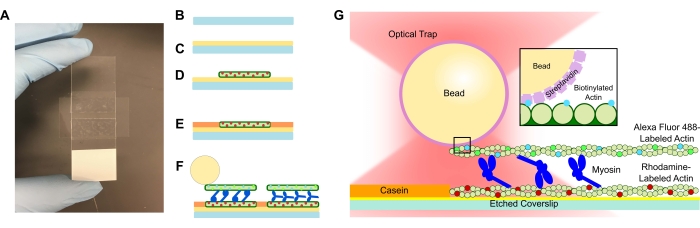
Figure 1: Assay schematic. (A–C) Etched coverslips are coated in poly-L-lysine and used to form the flow cell by using double-sided tape and a microscope slide. Timed introductions and incubation steps described in the protocol result in rhodamine-labeled phalloidin-stabilized actin as the template or bottom filament (D), followed by casein blocking to prevent non-specific binding (E), and (F) Alexa Fluor 488 phalloidin-stabilized biotinylated actin as the cargo or top filament, and teams of myosin II that slide the filaments apart and generate force when ATP is introduced. The geometry of the motors and nature of crosslinking within the bundle could vary under different conditions, such as salt concentration59. Previous studies have demonstrated that the myosin tail domain has the ability to interact with actin filaments and slow ensemble motility46. However, myosin heads in heavy meromyosin experiments demonstrate binding of each head to adjacent actin filaments60. (G) Streptavidin beads are used as the optical handle for the trap and bind solely to the cargo biotinylated actin filament, which aids in validating that proper bundles are formed on the slide. Please click here to view a larger version of this figure.
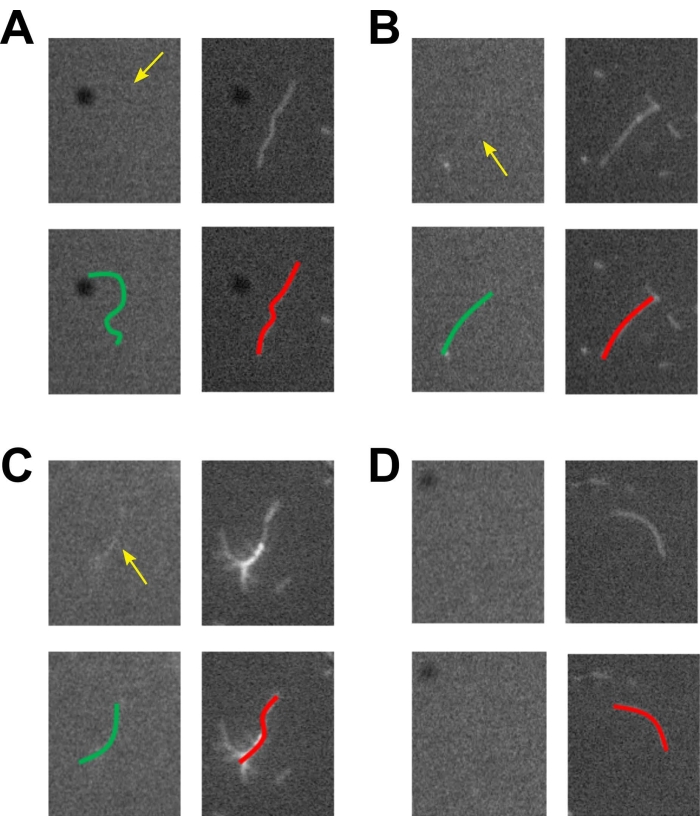
Figure 2: Fluorescent actomyosin bundles. Four different encounters of actin filaments and bundles within the bundle assay presented in Figure 1. The top cargo biotinylated actin filament with the Alexa Fluor 488 phalloidin channel is shown on the left, and the bottom template actin filament with the rhodamine phalloidin channel is on the right. At the bottom, the same figure is shown with colored lines overlaid to help guide the eye. (A) A top actin filament is found near a bottom actin filament but has an incomplete overlay. This would not be used for bundle experiments. (B) Top and bottom actin filaments are colocalized, and the intensity of each filament confirms that they are each single filaments within the bundle. This would be a good candidate for bundle experiments. (C) A large bundle of self-assembled rhodamine filaments is found on the bottom. While there is a corresponding top actin filament that is colocalized, there are too many bottom filaments present; thus, it would not be used for bundle experiments. This is also an example of how when multiple actin filaments of the same type are bundled, the fluorescence intensity increases. The user can utilize this as a gauge for judging single filaments versus bundles of the same filament type. (D) A bottom filament is present with no corresponding top filament, also confirming no bleedthrough. This would not be used for bundle experiments. We note that the intensity of the filaments in the Alexa Fluor 488 channel is low and believe it is due to the filter set that is being used (Filter Set 09 from Zeiss). The filter set used for the rhodamine channel is Filter Set 43 from Zeiss. Please click here to view a larger version of this figure.
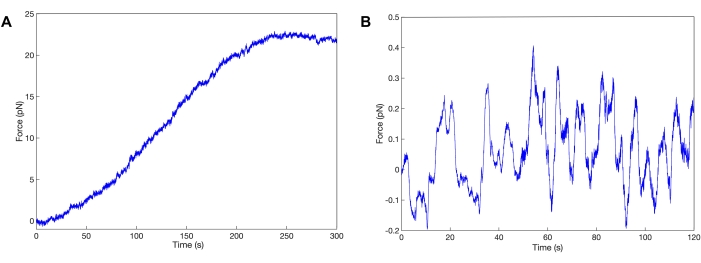
Figure 3: Myosin II ensemble force generation. Representative traces of skeletal myosin II motors generating force within the constructed in vitro actin structural hierarchy. The myosin motors are working together to collectively and productively generate force until a plateau is reached and force is sustained (A) or experience antagonization near baseline (B). Please click here to view a larger version of this figure.
Supplemental Figure S1: Bruker/JPK Nanotracker2 optical trap. (A) Computer monitor. (B) Computer keyboard. (C) Computer tower. (D) Controller box. (E) Laser power supply. (F) Optical trap optics box. (G) Inverted microscope. (H) Door to microscope stage. (I) Polarizer slider to switch between brightfield and differential interference contrast imaging. Please click here to download this File.
Supplemental Figure S2: Remote control for optical trap. (A) Keypad to position the motorized stage. (B–C) Adjust trap position. (D) A, X, and B switch on and off the main shutter, trap 1 shutter, and trap 2 shutter, respectively. (E) The Logitech button is used to wake up the controller. (F) The up and down buttons that are used to position the trapping objective. (G) The up and down buttons that are used to position the detection objective. Note that the remote control is not required, and all these manipulations can be accomplished in the software. However, it is convenient to be able to control the objectives and stage position while looking into the microscope stage environment. Please click here to download this File.
Supplemental Figure S3: Fluorescence module for optical trap. The 89North PhotoFluor fluorescence white light source is coupled to the back of the inverted microscope. It is turned on and off with a toggle switch (arrow). Please click here to download this File.
Supplemental Figure S4: Fluorescence filter cube turret. The turret (arrow) can be turned to use the filter cube necessary for imaging in DIC, rhodamine, or Alexa Fluor 488 dyes. Note that filter cubes can be switched out to customize the setup for using different fluorophores. Please click here to download this File.
Supplemental Figure S5: Nanotracker2 software. (A) Laser power button and control. (B) Objective positioning window. Directional arrows are used to move the detection (top) and trapping (bottom) objectives. Double arrows move the objectives at a higher speed. The blue and red button at the bottom-left uncouples the objectives and retracts them back to their original position. This is necessary for when taking samples in and out of the microscope stage. The third button from the left with the objectives and padlock icon "couples" the objectives so that when they are both in focus and achieve Kohler illumination, the user can move both the trapping and detection objectives up and down in the z-axis. (C) Sample positioning window used to move the microscope stage in the x- and y-axis. Double arrows move the stage at a higher speed. This window is activated by clicking the up/down and left/right arrow icon at the top menu. (D) Camera visualization window. The wrench icon can be used to set customized imaging conditions. This window is activated by clicking on the Camera icon at the top menu. (E) Microscope illumination window. This window is activated by clicking on the Light Bulb icon at the top menu. Please click here to download this File.
Supplemental Figure S6: Calibration window. (A)This window is used for bead calibration and is activated by clicking on the Cal icon at the top menu. To calibrate a bead, a best fit of the corner frequency is accomplished in the x, y, and z signals. (B) For each signal, choose the appropriate signal button in the top left. (C) Click on run and optimize the fit by clicking and dragging within the green window (D). (E) Once satisfied with the fit, click on Use It for sensitivity and stiffness. This will allow for recording displacement in nanometers and force in piconewtons. (F) Then, click on Accept Values at the bottom left. Repeat for the y and z directions. Please click here to download this File.
Supplemental Figure S7: Data Acquisition window. This window is used to acquire position and force data and allows the user to see the measurements in real time. (A) This window is activated by clicking the x,t icon at the top menu. (B) The user can switch between viewing the x and y signals. (C) Click on Start to begin visualizing data. Click on Autosave to save the data. Click on Start Record to begin recording and saving data. Please click here to download this File.
