It was hypothesized that the present in vitro differentiation (IVD) products would possess similar surface markers and anti-tumor abilities to human NK cells.
The defining marker of human NK cells was accessed to test whether the differentiation products from the 3D culture system yielded cells that phenotypically resembled human NK cells. About 15% of the cells dissociated from the 3D culture system from two batches induced at different time points (n = 2) were CD3−CD56+ (Figure 3A). CD3 was replaced with CD45 (a pan-leukocyte marker) in the test panel due to the absence of CD3 and the difficulty inducing T cells in vitro. Approximately 16% of the cells dissociated from the 3D structure were CD45+CD56+ (Figure 3B, n = 1), which is in line with the percentages of CD3−CD56+ cells seen in earlier trials. The percentages of CD3−CD56+ cells in the cells harvested from the 2D culture system ranged from 30% to 6%, from more successful inductions (Figure 3C, n = 3) to less successful inductions (Figure 3D, n = 2). The functionality and phenotypes of the cells harvested from the 2D culture system were validated. During the Day 18 culture from the 2D system, about 12% of the cells expressed both ectopic CD56 and CD107a after 2 h of 50 ng/mL IL-18, 455 IU/mL IL-2, and 20 ng/mL anti-CD244 antibody stimulation (Figure 4C, n = 1). About 28% of the harvested cells were CD94+CD159a+ (Figure 4B, n = 1). The ectopic expression of CD107a, a protein lining on the membrane of granules, indicates degranulation10, while the CD94/NKG2A(CD159a) heterodimer is another defining marker of NK cells. This provides an example of the phenotypic and functionality validation of IVD products.
The functionality of the IVD products was tested by their cytotoxicity against human erythroleukemia cells-K562 cells. Comprehensive ligand profiling on K562 cells reveals their downregulated major histochemistry complex I (MHC-I) and comparatively strong NKG2D ligand expression (such as ULBP-1, ULBP-2/5/6, and ULBP-3)11; thus, K562 cells are susceptible to the lytic activity of NK cells12. Both IVD endpoint products and NK92mi cells were primed with 50 ng/mL IL-18, 455 IU/mL IL-2, and 20 ng/mL anti-CD244 antibodies overnight. The GFP+ K562 cells were cocultured with either harvested cells from IVD or NK92mi cells at different effector-to-target ratios (E:T) in the presence of 50 ng/mL (IU not provided) IL-18, 455 IU/mL IL-2, and 20 ng/mL anti-CD244 antibodies in the same volume of H5100 medium for 3 h. The readout of NK cell killing activity was the expression of the dead cell marker on GFP+ subsets. The K562 cells were transduced with a commercially available control vector (see the Table of Materials) to express GFP constitutively, and the efficiency of the transduction was about 80% (Supplementary Figure 1A). The percentages of GFP signal added were proportional to the number of GFP+ K562 cells added, suggesting specific GFP expression from the transduced K562 cells (Supplementary Figure 1B).
The percentages of dying target cells shown in Figure 5 were calculated with the following formula13:
Percentage of dying cells = (FITC + DAPI + % of coculture [for example, Figure 5A]) – (FITC + DAPI + % of transduced K562 cells)
Of all three E:T ratios assessed (0.1:1, 0.33:1, 1:1), the percentages of dying GFP+ cells were positively correlated with the E:T ratios when cocultured with IVD products (Figure 5B, hEPSC-NK) or NK92mi cells (Figure 5B, NK92mi), indicating the specific killing of tumor targets. However, the cytotoxicity of the IVD products was comparatively modest within the tested timeframe (Figure 5C).
Finally, the lymphoid progenitor cell generation was compared between the 3D and 2D cultures. It was found that the 3D culture condition generated more progenitor cells (55,000 cells) than the 2D culture condition (36,000 cells) (Supplementary Figure 2). This suggests that the context of tissue architecture and cellular components in the 3D culture supported the generation of lymphoid progenitor cells.
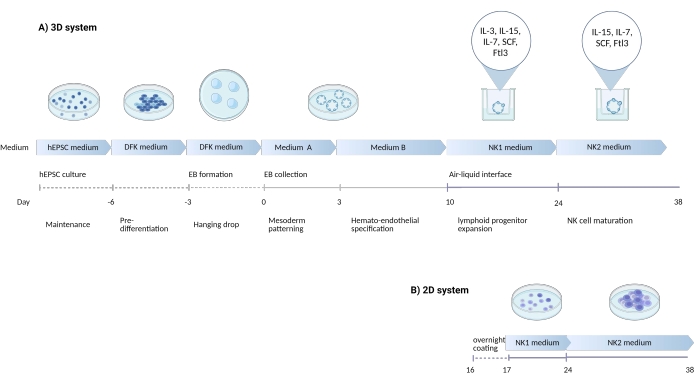
Figure 1: Schematic diagram showing the differentiation strategy to differentiate NK cells from hEPSCs. (A) Here, the timeline of the 3D culture systems is shown, detailing the culture conditions and brief keywords from each step. The air-liquid interface of the 3D system is maintained during the NK cell induction. (B) Here, the timeline of the 2D culture systems is shown. The released cells harvested from Days 7-10 from structures in the 3D system are seeded on a coated plate without the air-liquid interface. Please click here to view a larger version of this figure.
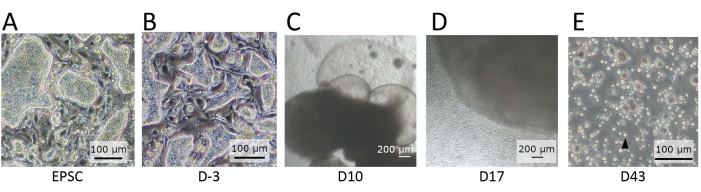
Figure 2: Morphology of the cells observed at distinct stages of differentiation. (A) The morphology of hEPSCs when maintained in EPSCM. The hEPSCs form domed-shaped colonies. (B) The morphology of hEPSCs after pre-differentiation. The domed-shaped colonies are flattened. The hEPSCs are harvested and form EBs with the hanging drop technique. The EBs are collected and cultured in medium A and then medium B. (C) The morphology of an EB on Day 10. During this period, the EB starts to expand and inflate, forming membranous structures. (D) The morphology of a cell-releasing EB. After seeding on the transwell, the EB grows and constantly releases round cells to the surroundings. Some of the released cells are collected and cultured on a 12-well plate coated with lymphoid differentiation coating materials. The cell populations are morphologically heterogeneous and tend to aggregate together. (E) The morphology of cells observed at the endpoint of the IVD. Small, circular suspension cells (black arrow) are observed. Scale bars: (A,B,E) = 100 µm; (C,D) = 100 µm. Please click here to view a larger version of this figure.
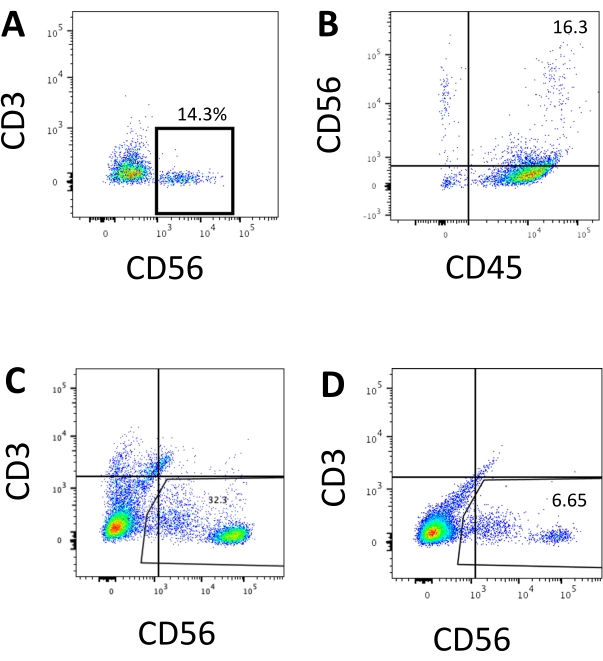
Figure 3: Representative FACS plots of hEPSC-derived products at the endpoint. Cells are incubated with fluoroscope-conjugated antibodies on ice for 30 min. The samples are stained with DAPI before being loading into the FACS sample injection port. Unstained cells are used to establish the negative gating. (A) Representative FACS plot on CD3 and CD56 expression in cells dissociated from the 3D culture system from two batches (n = 2). (B) Representative FACS plot on CD45 and CD56 expression in cells dissociated from the 3D culture (n = 1). (C) Representative FACS plot on CD3 and CD56 expression in cells harvested from the coated plate from a successful induction (n = 3). (D) Representative FACS plot on CD3 and CD56 expression from the coated plate when the induction is suboptimal (n = 2). Please click here to view a larger version of this figure.
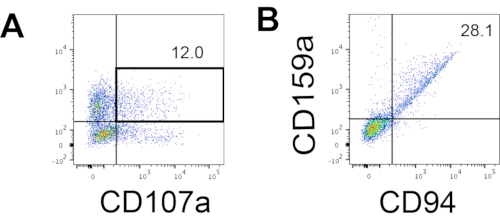
Figure 4: Representative FACS plot on hEPSC-derived products during Day 18 culture (n = 1). (A) FACS plot on CD56 and CD107a expression after 2 h of stimulation with IL2, IL-18, and anti-CD244 antibodies. (B) FACS plot on CD159a and CD94 expression. The samples are stained withDAPI before being loaded into the FACS sample injection port. Unstained cells are used to establish the negative gating. Please click here to view a larger version of this figure.
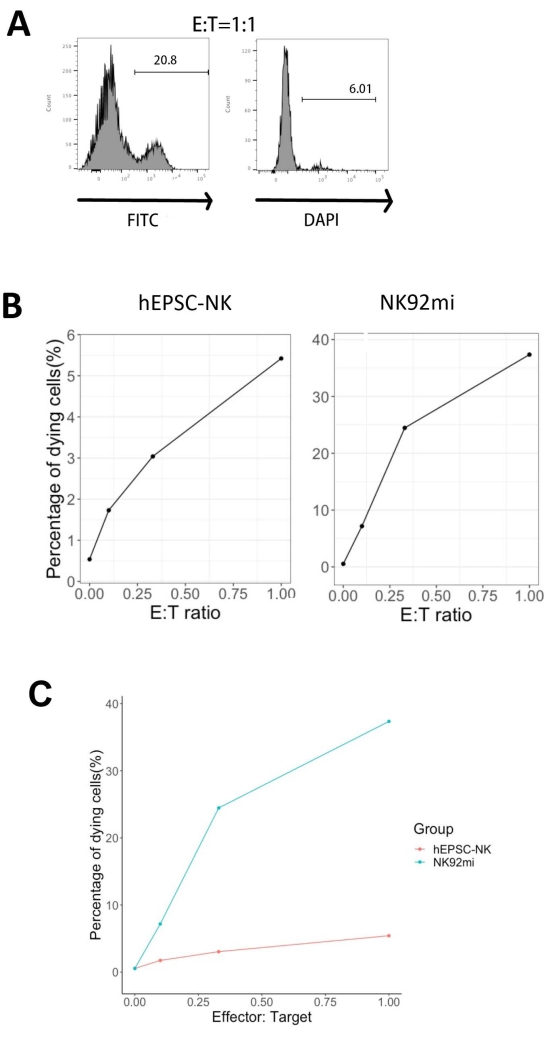
Figure 5: Plot of the percentages of dying cells against different E:T ratios from the coculture of GFP+ K562 cells with either the IVD endpoint product or NK92mi cells (n = 1). The expression of FITC+ DAPI+ cells was assessed with a cell analyzer, and the data analysis was performed with compatible software. (A) A representative FACS plot showing the gating of FITC + DAPI + subsets from the coculture of IVD products and K562 cells with an E:T ratio of 1. (B) Individual plots from the coculture experiment with GFP+ IVD products (left) or NK92mi cells (right) for 3 h. Both reflect a positive proportional relationship between the percentage of dying cells and the E:T ratio. (C) A plot showing the killing curves of IVD products and NK92mi cells on the same scale. The IVD products displayed mild cytotoxicity as compared to the NK92mi cells. Please click here to view a larger version of this figure.
Supplementary Figure 1: Functional assay of NK cells from hEPSCs. (A) FACS histogram of transduced K562 cells. The FITC+ K562 cells expressed GFP. (B) FACS histograms from the coculture of GFP+ K562 cells and the endpoint IVD products at three E:T ratios (n = 1). The percentages of FITC+ cells in the coculture corresponded to the number of added transduced K562 cells. The gatings were established with untransduced K562 cells. Please click here to download this File.
Supplementary Figure 2: Lymphoid progenitor differentiation in 2D versus 3D culture. The present organoid-based protocol was used to induce lymphoid progenitors from human expanded potential stem cells. Two conditions were set up: (1) lymphoid progenitors were kept in culture within the organoids (3D), and (2) lymphoid progenitors were isolated and kept on the culture dish (2D). After 2 weeks of additional culture, the cell numbers were determined to compare the 2D versus 3D culture. Please click here to download this File.
