MitoTracker Green is a green-fluorescent mitochondrial stain that is able to accurately localize to mitochondria. The dye can easily stain live cells and is less effective in staining aldehyde-fixed or dead cells (Figure 2). The red-fluorescent lysosome dye LysoTracker Red is capable of labeling and tracking acidic lysosomal organelles and can only stain live cells (Figure 2). Confocal microscope imaging allows the visualization of mitochondria and lysosomes stained with the appropriate dyes (Figure 1 and Figure 2).
Mitophagy is a catabolic cellular process that selectively degrades mitochondria by autophagy, which usually occurs in damaged mitochondria after injury, aging, or stress20. Subsequently, these mitochondria are delivered to lysosomes for degradation. Mitophagy helps maintain the quantity and quality of mitochondria in a healthy state in a wide range of cell types. In healthy mammalian cells, mitophagy occurs infrequently, and therefore other stimuli are required to induce this process25. Carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP), a mitochondrial uncoupler, is a nonspecific ionophore that causes a severe loss of mitochondrial membrane potential within minutes, alteration of intracellular pH, and subsequent mitophagy26,27. In this study, FCCP was used to trigger mitophagy in MEF cells for confocal imaging. When damaged green-stained mitochondria are engulfed by red-stained lysosomes, the green and red fluorescence overlap to reveal yellow co-localized mitochondria-lysosomes (Figure 3). The yellow dots in Figure 3D and Figure 4B correspond to these co-localized mitochondria-lysosomes, representing ongoing mitophagy, and thus can be counted to evaluate the extent of mitophagy (Figure 4).
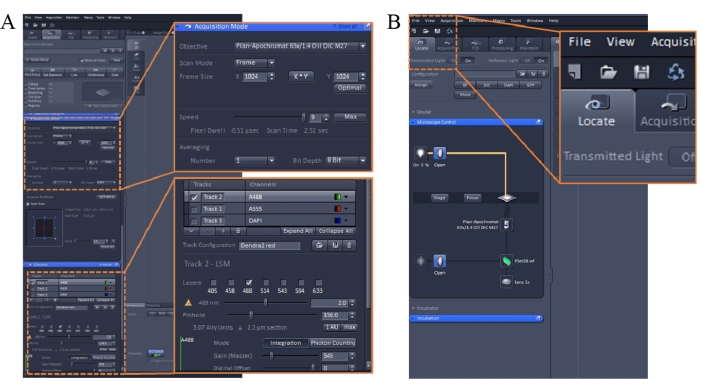
Figure 1: Imaging parameters of the confocal microscopy imaging software. Please click here to view a larger version of this figure.
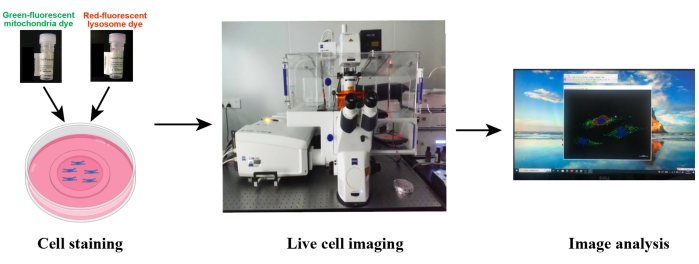
Figure 2: Schematic illustration of the confocal imaging of live cells. The live cells are co-stained with the green-fluorescent mitochondrial dye and red-fluorescent lysosomal dye, and then the live cells are imaged using confocal microscopy. Image processing and data analysis was performed using the microscope-associated imaging software or Image J. Please click here to view a larger version of this figure.
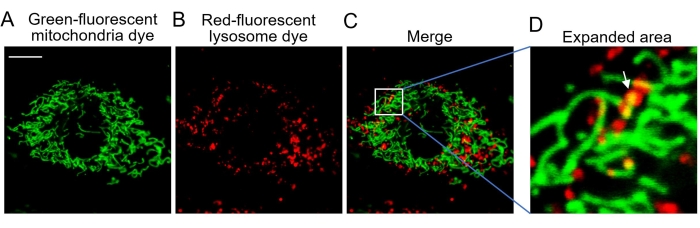
Figure 3: Confocal imaging of live cells. (A) Representative images of cells stained with the green-fluorescent mitochondria dye show the mitochondria. (B) Representative images of cells stained with the red-fluorescent lysosome dye showing the lysosome. (C) Merged image of both fluorescent dyes. (D) Expanded area showing mitophagy. The white arrow indicates green mitochondria engulfed by red lysosomes. Abbreviation: Ex = excitation wavelength. The green-fluorescent mitochondria dye is excited at 488 nm with emission collected at 505-545 nm. The red-fluorescent lysosome dye is excited at 543 nm with emission collected at >560 nm. Scale bars = 10 µm. Please click here to view a larger version of this figure.
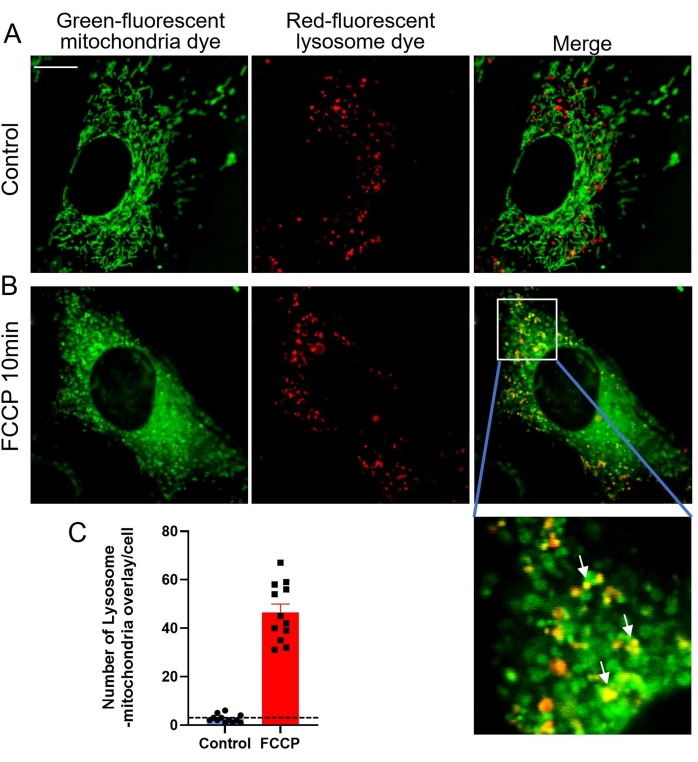
Figure 4: Mitophagy triggered by FCCP stimulation. (A) Representative images of cells co-stained with the green-fluorescent mitochondria dye and the red-fluorescent lysosome dye. (B) Representative images of cells treated with 1 µM FCCP for 10 min. The white arrow indicates green mitochondria engulfed by red lysosomes. (C) Quantitative data of mitophagy indicated by lysosomal-mitochondria overlay. Data are mean ± SEM, n = 8 cells. *p < 0.05 versus control. Scale bars = 10 µm. Please click here to view a larger version of this figure.
