Ensaio para Morte Celular: Ensaio de Liberação de Cromo para Avaliar a Capacidade Citotóxica
English
Condividere
Panoramica
Fonte: Frances V. Sjaastad1,2, Whitney Swanson2,3e Thomas S. Griffith1,2,3,4
1 Programa de Pós-Graduação em Microbiologia, Imunologia e Biologia do Câncer, Universidade de Minnesota, Minneapolis, MN 55455
2 Centro de Imunologia, Universidade de Minnesota, Minneapolis, MN 55455
3 Departamento de Urologia, Universidade de Minnesota, Minneapolis, MN 55455
4 Centro de Câncer Maçônico, Universidade de Minnesota, Minneapolis, MN 55455
Uma das principais funções das células do sistema imunológico é remover células-alvo que foram infectadas com vírus ou passaram por transformação em células tumorais. Ensaios in vitro para medir a capacidade citotóxica das células imunes têm sido um grampo em laboratórios por muitos anos. Esses ensaios têm sido usados para determinar a capacidade de células T, células NK ou qualquer outra célula imune para matar células-alvo de forma específica de antígeno ou -inespecífica. Ligantes de morte (por exemplo, ligante fas ou TRAIL), citocinas (por exemplo, IFNg ou TNF), ou grânulos citotóxicos (ou seja, perforina/granzyme B) expressos por células effectoras são algumas maneiras pelas quais a morte celular alvo pode ser induzida. Com a explosão na pesquisa de imunoterapia tumoral nos últimos anos, há um crescente interesse em encontrar agentes para aumentar a atividade citotóxica das células imunes para melhorar os resultados dos pacientes. Por outro lado, algumas doenças são marcadas pela atividade excessivamente exuberante da atividade citotóxica das células imunes, resultando em esforços para identificar agentes para temperar essas respostas. Assim, ter um ensaio no qual o usuário pode facilmente integrar qualquer número de células efeitos, células-alvo e/ou modificadores de resposta no design experimental pode servir como um meio valioso de avaliar rapidamente a capacidade citotóxica das células effectoras e/ou a capacidade de resposta da célula alvo.
Esses ensaios in vitro envolvem a mistura de diferentes populações celulares, bem como o uso de um número relativamente baixo de células-alvo e efeitos. Assim, uma necessidade do ensaio é rotular as células-alvo de uma maneira que possa ser facilmente detectada e quantitada, permitindo ao usuário determinar então a “por cento de lise específica” mediada pelas células effectoras. A radioatividade – especialmente, o cromo 51 (51Cr) na forma de Na251CrO4– é uma maneira barata de rotular rapidamente e sem especificação proteínas celulares dentro das células-alvo (1). Os tempos de rotulagem curta e ensaio total reduzem o potencial de mudanças significativas no número e/ou fenótipo das células-alvo, o que poderia influenciar o resultado do ensaio. Após a perda da integridade da membrana das células-alvo como resultado da atividade citotóxica das células effectoras, as proteínas celulares rotuladas por Cr de 51Cr dentro das células-alvo são liberadas na cultura sobrenante, tornando-se disponíveis para quantitação. Como em qualquer ensaio que examine a função das células imunes in vitro,há uma série de considerações importantes para considerar melhorar o desempenho do experimento. Uma das características mais críticas é usar células de efeito saudável (para atividade citotóxica máxima) e alvo (para resposta máxima e morte espontânea mínima/ liberação de51Cr). É necessário contato com o efeito e a célula alvo (levando ao uso comum de placas de fundo redondo de 96 poços para incentivar o contato celular) (2). Por fim, a análise dos dados depende da inclusão de populações celulares alvo de controle positivo e negativo.
O protocolo a seguir delineará as etapas para a realização de um ensaio de liberação padrão de 51Cr para medir a capacidade citotóxica de uma população de células efeitos, embora uma versão nãoradioativa usando europium tenha sido recentemente desenvolvida. 51 Cr é um poderoso emissor de radiação γ. Consequentemente, o uso deste ensaio requer treinamento adequado de segurança de radiação, espaço de laboratório dedicado, um contador gama e descarte de amostras radioativas.
A sequência geral de eventos neste ensaio são: 1) preparar 51alvos rotulados por Cr; 2) preparar células de efeitos e adicionar à placa enquanto as células-alvo estiverem rotulando; 3) adicionar alvos rotulados à placa; 4) placa incubadora; 5) supernastas de colheita; e 6) analisar dados após a execução de amostras no balcão. As amostras são comumente preparadas em triplicado, e então mediadas para explicar quaisquer diferenças sutis de pipetação.
O EPI adequado é importante para este ensaio. Especificamente, o usuário deve usar um jaleco e luvas. Os óculos de segurança podem ser necessários com base no laboratório ou instituição. Deve haver ampla proteção de chumbo para armazenamento seguro e uso do 51Cr durante todas as etapas. Por fim, deve haver espaço de laboratório dedicado e equipamentos reservados para o uso de 51Cr, incluindo toda a sinalização adequada para indicar onde as amostras com 51Cr estão sendo mantidas e um contador Geiger equipado com sonda gama para examinar o espaço para possível contaminação.
Neste exercício de laboratório, determinaremos a capacidade das células mononucleares de sangue periféricos humanos (PBMCs), (CpG estimuladas vs. não estimuladas) para matar células de melanoma, usando a linha celular de melanoma humano WM793 como modelo e o ensaio de liberação de cromo.
Procedura
Risultati
In this example, effector cells stimulated with CpG (Figure 1, black circles) killed the target cells more effectively, as the ratio of effector cells to target cells increased. This increase was not observed in the unstimulated PBMCs (white circles), indicating that CpG stimulation is necessary for the observed increase in target cell lysis.
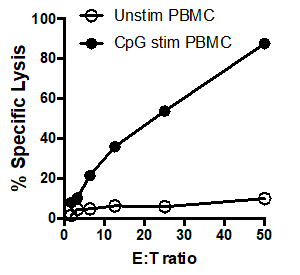
Figure 1: 51Cr assay scatter plot: Tumoricidal activity by human PBMCs, unstimulated (white circles) and after stimulation with CpG (black circles), tested at different effector: target cell ratios (E: T) ratios (ranging from. 50:1 to 1.5:1).
Applications and Summary
The assay described here has considerable flexibility, as a variety of effector and target cells can be used depending on the question being asked. For example, effector cell specificity can be determined by using different target cells or the mechanism of effector cell killing can be determined by using cells deficient in specific proteins or using protein specific inhibitors. A major problem with the 51Cr release assay is the potential for a high spontaneous release rates by the target cells. When cultured alone (without effector cells), the spontaneous release of 51Cr by the target cells should ideally be no more than 30% of the total ("maximal") release by the target cells immediately lysis. Higher spontaneous release rates may be due to using unhealthy target cells, either due to poor health (e.g., extended culture of a cell line) or an overly long labelling period.
Riferimenti
- Brunner, K. T., Mauel, J., Cerottini, J. C. and Chapuis. B. Quantitative assay of the lytic action of immune lymphoid cells on 51Cr-labelled allogeneic target cells in vitro; inhibition by isoantibody and by drugs. Immunology, 14 (2):181-196, (1968).
- Kemp, T. J., B. D. Elzey, and T. S. Griffith. Plasmacytoid dendritic cell-derived IFN-alpha induces TNF-related apoptosis-inducing ligand/Apo-2L-mediated antitumor activity by human monocytes following CpG oligodeoxynucleotide stimulation. The Journal of Immunology, 171 (1): 212-218, (2003).
Trascrizione
In this video you will observe how to perform the chromium release assay and determine the cytotoxic potential of the effector cells.
Immune cells are responsible for identifying and removing potentially harmful cells, like cancer or virus-infected cells, from the body, which is an integral part of the immune response. Several immune cells, like T-cells and NK cells, possess a property known as cytotoxic potential, which is the ability to identify target cells and secrete proteins that induce protein degradation, lysis, and death of those target cells. Quantifying cytotoxic potential is critical for measuring immune cell activation and potency, and the chromium release assay is commonly used for this purpose.
This method enables users to compare cytoxicity induced by specific types of immune cells under different conditions, which is valuable for studying cancer immunotherapy and immunity related diseases. To begin, the target cells, like cancer cells, are incubated with a radioactive isotope, chromium 51, which is taken up by the cells. Next, these radio labeled cells are co-cultured with the isolated immune cells of interest, also called the effector cells, in a round bottom, 96- well plate to facilitate interaction between the two cell types.
The overall setup of the assay involves incubating a specific number of target cells with different concentrations of the immune cells, along with appropriate controls. The co-culture allows the effector cells to induce apoptosis and lysis in the target cells, resulting in the release of the intracellular chromium 51 into the supernatant. Then, at a preoptimized time point, the supernatant containing the released chromium is harvested from all the wells. The chromium 51, being radioactive, spontaneously undergoes radioactive decay to emit gamma radiation. The gamma radiation levels in the supernatants from all the wells in the assay plate represent a quantifiable output of the lysis of the target cells. This is measured using a gamma counter, which is then used to determine the cytotoxic potential of the immune cells.
To begin, the target cells, human melanoma cell line WM793 in this example, are prepared into a single cell suspension. To do this, first remove the media from the tissue culture flask and wash the cells with five milliliters of 1X PBS. Decant the PBS and then add one milliliter of trypsin to the plate for approximately two minutes. Gently tap the flask to loosen the cells from the flask surface and then add five milliliters of RPMI media to the flask. Pipette the media up and down to collect the cells and add this suspension to a 15 milliliter conical tube.
Place the tube in the centrifuge for five minutes at 1200 RPM. Next, remove the media from the tube making sure not to disrupt the cell pellet. Gently flick the bottom of the tube to disrupt the cell pellet and add 10 milliliters of media to the tube. Then, gently pipette the media up and down to bring the cells into suspension. Next, determine the cell concentration using a hemocytometer and transfer two milliliters of the original cell suspension to a new 15 milliliter conical tube. Place the tube into a centrifuge and pellet the cells at 12 hundred RPM for five minutes. After centrifugation, pour the excess media out of the tube into a waste container. Briefly vortex the tube to resuspend the cell pellet in the small volume of medium left behind.
Next, prepare to use Chromium 51 by moving to a lab space dedicated for this particular radioactivity. There should be ample lead shielding for safe storage and use of the Chromium 51 during all steps, as well as proper signage to indicate where samples with Chromium 51 are being kept. A Geiger counter equipped with a pancake probe is also necessary to serve in the space for possible contamination.
Once set up for the proper use of radioactivity, add 100 microcuries of Chromium 51 directly to the target cell suspension. Then, add a small piece of radioactive tape to the tube to indicate that the sample and tube are now radioactive. Place the tube in a 37 degree celsius incubator with a lead shield and incubate for an hour, flicking the tube every 15 to 20 minutes.
While the target cells are labeling, prepare a single cell suspension of effector cells. In this example, human peripheral blood mono nuclear cells, or PDMCs, were isolated from whole blood by standard density gradient centrifugation to a concentration of 5 times 10 to the 6th. Transfer this effector cell suspension into a disposable reagent reservoir and then add 200 microliters of this suspension into each well of row B in a 96-well round-bottom plate. Next, add 100 microliters of RPMI to each well in row C through G of the plate.
Now, begin performing serial dilutions of the PBMCs to have a range of effector cell numbers by first removing 100 microliters of the cells in the wells in row B and adding this to row C. Then, further dilute the effector cells by transferring 100 microliters of cells from row C to row D. Continue the serial dilution. Once row G is reached, move 100 microliters from the wells to leave a final volume of 100 microliters in each well in that row. Next, add 100 microliters of tissue culture medium to the wells in row A to serve as a control for the spontaneous release of Chromium 51 from the target cells, as no effector cells should be added to this row. Then, place a plate into a 37 degree celsius incubator until the target cells are ready to be added.
After the incubation period, remove the target cells from the incubator and wash with 5 milliliters of FBS to remove any excess Chromium 51. Then, place the tube in a designated centrifuge and spin at 1200 rpm for 5 minutes. Remove the radioactive FBS wash into an appropriate waste container and repeat the wash step by resuspending the pellet in a fresh 5 milliliters of FBS. Place the tube in a designated centrifuge and spin the cells again at 1200 rpm for 5 minutes. Remove the second wash and check the pellet for incorporated radioactivity using a Geiger counter. Finally, Resuspend the pellet in 10 milliliters of complete medium and pour the Chromium 51 labeled, target cell suspension into a disposable reagent reservoir. Then, add 100 microliters of these labeled target cells to every well of the 96-well effector cell plate. Next, add 100 microliters of 1% NP-40 in water to the wells in row H to lyse all the target cells this each row. These wells will be used as a control to determine the total counts per minute, or cpm.
Now that the plate is prepared, secure the lid by adding a small piece of tape to the each side of the plate and place a piece of radioactive tape on the lid to indicate it contains chromium 51. Then, place the plate in a centrifuge marked to handle radioactive samples. If only one experimental plate is being used, add a balance plate to the centrifuge. Set the centrifuge to 1200 rpm, and bring the plate up to speed. Once at the speed, stop the machine. Remove the plate from the centrifuge. Then, place the plate in a 37 degree celsius incubator with a small piece of lead shielding over the plate for additional safety. Incubate for 16 hours to allow the target cells to lyse.
At the end of the incubation period, carefully remove the tape around the edge of the plate, and remove the lid. Next, place the harvesting frame on the plate making sure to confirm the small filter discs are in place for each of the cotton plugs. Now, slowly and gently press the cotton plugs into the wells. After approximately ten seconds, release the pressure on the cotton plugs, and then transfer the cotton plugs to tube strips. Place each of these tubes into a secondary FACS tube. Finally, load the FACS tubes onto a gamma counter and run the samples to quantitate the amount of chromium 51 released in each condition. Carefully record the order in which the tubes were loaded into the counter.
Here, unstimulated PBMCs were added to the first 3 lanes and CPG stimulated PMBCs were added to lanes 4 through 6. In this example, the counts per minute were entered into the cells of a spreadsheet in the same manner as the samples were laid out in the original plate and the averages of the triplicates were calculated. For example, for the first condition, cells A1, A2, and A3 were averaged in cell I3. Once the averages are determined, the percent of specific lysis for each condition can be calculated using this formula. For example, to calculate the percent specific lysis for the unstimulated cells that had a ratio of 50 to 1 effector cells to target cells the spontaneous CPM, which in this example, is 1164.67, was subtracted from the experimental CPM, 1129. 67. This number can then be divided by the difference between the maximum CPM and the spontaneous CPM, and then multiplied by 100 to give the percent specific lysis. This is then calculated for each condition. These data can then be graphed to show comparison of the E to T ratio with the percent specific lysis for both the unstimulated PBMCs, and the CPG stimulated PBMCs. In this example, effector cells stimulated with CPG more effectively killed target cells as the ratio of effector cells to target cells increased. This increase was not observed in the unstimulated PBMCs, indicating that CPG stimulation is necessary for the observed increase in target cell lysis.