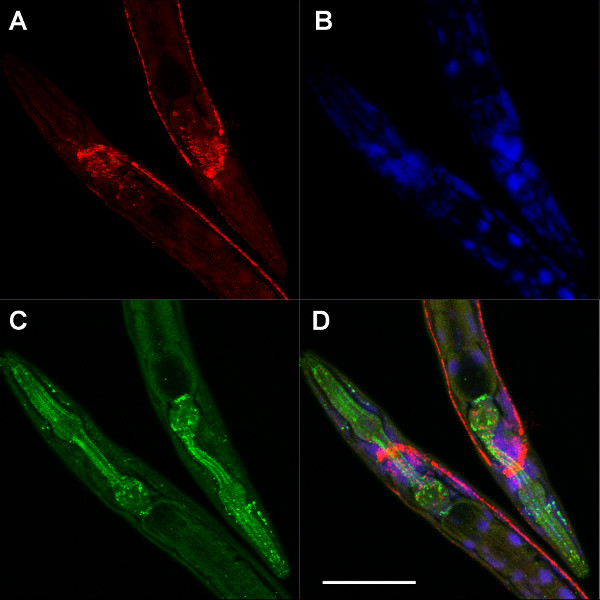
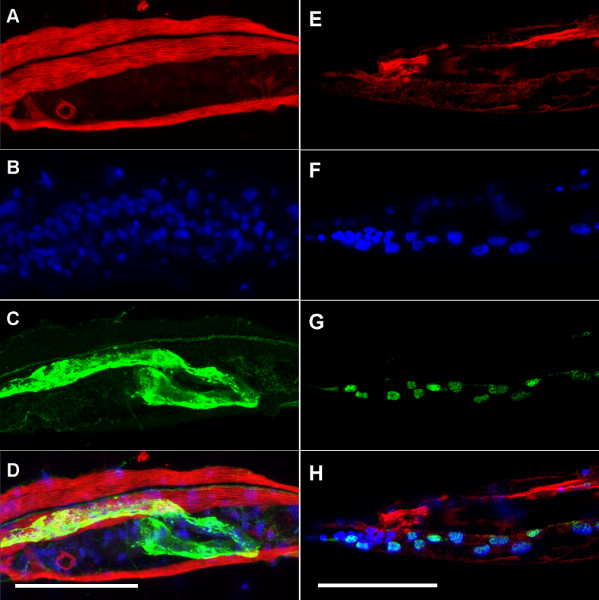
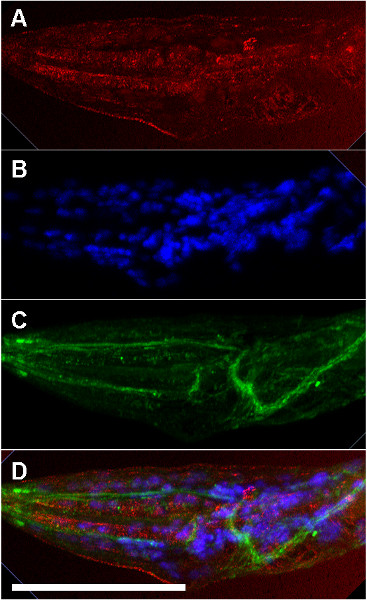
NOTE: Multiple protocol steps are presented with options for set-up and preparation depending on the specific conditions of the experiment. For these cases, alternative steps are presented as A, B, C, etc. The protocol with alternative steps is outlined in Figure 1.
1. Preparation of Polylysine Coated Slides
Three different types of slides may be used, depending upon the desired trade-off between ease of preparation, relative adhesion, and non-specific binding of the antibody. Details of slide preparation alternatives are given below in steps 1A-1C in order of increasing adhesion and complexity. Slides prepared by these different methods can be used together for a single experiment.
1A. No slide preparation
Commercially available polylysine coated slides provide low adhesion and low background.
1B. Slide preparation optimum for methanol-acetone fixation
Medium adhesion and low background.
- Prepare a polylysine solution for dipping the slides: Add 400 mg of very high molecular weight (150,000 – 300,000 D) poly-L-lysine to 200 ml double distilled water. Add 2 ml 10% sodium azide stock (final concentration 0.1%) as a preservative. CAUTION: dry sodium azide is reactive and all forms are toxic. Wear mask and gloves while weighing powder to make 10% stock in double distilled water.
- Wipe single end frosted glass slides with lint-free wipes, removing smudges and particles. This should be done even on slides purchased with the designation "precleaned."
- Place slides back-to-back in a slide holder, coplin jar or other slide staining container, keeping frosted edges up and not quite perfectly aligned (to make later separation easier). Place slice holder in beaker with 70% ethanol (reagent grade alcohol not needed) with 1% HCl in distilled water or fill staining jar to level of frosting. Rock slides in solution for a minimum of 5 min. CAUTION: This solution is corrosive, wear gloves. NOTE: This solution can be reused several times (up to several months).
- Rinse slides in running distilled water for 1 min.
- Dry slides completely in a dust free environment either by placing in a 40-60 °C oven for 30-60 min or by keeping overnight at room temperature.
- Incubate slides in polylysine solution (covering unfrosted parts) on rocker for 15 min.
- Repeat drying of slides.
- Repeat polylysine solution incubation and drying steps.
- Separate the slides using a slim metal object such as a thin weighing spatula. If they are properly adhered, they are difficult to separate. Wear appropriate safety gear (safety glasses) and work on bench top paper (to be discarded after use), since small bits of glass may fly loose when slides are separated.
- Store slides in clean (dust-free) slide box at 4 °C until needed.
1C. Slide preparation optimum for formaldehyde fixation
Highest adhesion but high background.
- Place slides prepared with method 1B flat in a dust-free, oven-safe dish (for drying in oven) or on a flat dust free surface (for air drying).
- Place a 20-60 μl drop of the polylysine solution on the middle of each slide.
- Dry slides completely in oven or at room temperature. The polylysine will leave behind pale ovals as it evaporates. These ovals are very adhesive. Unfortunately, they also bind to the primary and secondary antibodies, even after blocking.
- Store slides in clean (dust-free) slide box at 4 °C until needed.
2. Preparation of Frozen Nematode Slides
Prepare nematodes for staining by completely rinsing free of bacteria using method 2A (for plates of nematodes) or 2B (for individual nematodes).
2A. Preparation of slides from plates of nematodes
- Place a flat piece of metal on dry ice to prechill.
- Use distilled water and a glass pipette (decreases loss of worms) or micropipette to rinse worms from NGM plate with bacteria into a 1.5 ml microfuge tube. Plates can be synchronized or mixed ages; do not use plates with many dauers (difficult to crack) or starved worms (increased autofluorescence) unless necessary.
- Rinse the worms 3-4x with distilled water until free of bacteria. (To decrease loss of worms use the same glass pipette.) To pellet worms, either let them sit on the bench for 3-5 min (to collect mostly adults at bottom of tube) or spin 30 sec at ~1,000 rpm (~500 g) in a tabletop centrifuge (to collect adults, larva, and embryos).
- Remove most of water from tube with worms, leaving about twice the volume of water as worms (but at least 25 μl total volume).
- Label frosted side of the "bottom" (treated in step 1) polylysine slides with indelible ink and place slides on counter next to container with dry ice, label side up.
- Have an equal number of regular (wiped clean but not polylysine treated) "top" slides available.
- Place ~25 μl worms in water on the "bottom" adherent slide and spread the liquid containing the worms over the central portion of the slide using the side of pipette tip.
- Let the worms settle for 30 sec to 2 min. If the slide is appropriately sticky, the ends of the worms will stick as they contact the slide.
- Hold the bottom slide in one hand and place the top slide over the bottom slide so that all but the frosted parts are overlapping. The liquid should keep the slides slightly apart, so that the worms still move slightly. Do not let the slides slip over one another – this twists the worms.
- Carefully press straight down slightly on the top slide, using the thumbs and forefingers of each hand. With the ideal amount of pressure, a few of the largest hermaphrodites on the edge of the slides will rupture, while most of the adults and larvae will contact each slide but will stay intact.
- Immediately and carefully (without slipping) put the slides on the piece of metal on dry ice. The slides should appear frozen within one minute. Keep on dry ice for 5 min until thoroughly frozen.
- At this point, slides may be stored in a labeled box at -80 °C for several days. (If they are kept weeks at -80 °C, the water will start to sublime away and the worms will not stain as well). If slides are stored at -80 °C, let them 'warm up' on dry ice for 10 min before cracking.
- Proceed to step 3 (fixation).
2B. Preparation of slides of individual nematodes
- Place a flat piece of metal on dry ice to prechill.
- Use a worm pick to transfer individual nematodes to a drop of water on an NGM plate to free them from bacteria. Nematode(s) should be well-fed to decrease autofluorescence, if possible. Repeat transfer to new spots of water as needed until worm(s) are free of bacteria.
- Label frosted side of the "bottom" polylysine slides with indelible ink and place slides on counter next to container with dry ice, label side up. Have an equal number of less adherent "top" (untreated) slides available.
- Place a 5-10 μl drop of water on the region of the bottom slide double-treated with polylysine. Use the larger volume if transferring more worms, to prevent the water from evaporating before completion.
- Transfer the worm(s) to the drop of water with the worm pick, using the microscope to watch as the worms sink down to touch the sticky slide surface. Transfer as few as one worm to as many as 20+ worms per drop.
- Hold the bottom slide in one hand and place the top slide over the bottom slide so that all but the frosted parts are overlapping.
- Immediately and carefully (without slipping or compressing) put the slides on the piece of metal on dry ice. Keep on dry ice for 5-30 min. (Do not store these slides long term, as the slides easily dry out.)
- Proceed to step 3 (fixation).
3. Fixation
Fixatives are necessary to 'fix' the antigen in place in the cell, by either precipitating ("light fixation") or cross-linking ("hard fixation") the antigen. Fixation may disrupt antigenicity; most known antibodies work best with a particular fixation condition. For new antibodies, a range of fixation conditions should be tested.
For any fixation, the first step is to make phosphate buffered saline solution. Next fix slides with nematodes for antibody staining using method A (light fix using methanol and acetone) or B (harder fix using formaldehyde or glutaraldehyde).
- Prepare a sterile 10x PBS (phosphate buffered saline) stock solution by adding 80 g NaCl, 2.0 g KCl, 27.2 g Na2HPO4•H20, 2.4 g KH2PO4, 20 ml 10% sodium azide stock. To make this stock solution, weigh out sodium azide powder to make 10% solution in double distilled water. Stir saline with gentle heat until dissolved. CAUTION: dry sodium azide is reactive and all forms are toxic. Wear mask and gloves when working with dry sodium azide or solutions.
- Let saline cool (Tris pH is temperature sensitive), then pH to 7.2 with 1-10 M NaOH. Top to 1 L with double distilled water and autoclave to sterilize. Store at room temperature and dilute to 1x as needed with sterile double distilled water. CAUTION: NaOH is caustic – wear gloves during use.
3A. Light fix using methanol-acetone
- Put 100% methanol, 100% acetone, and 1x phosphate buffered saline (PBS) in staining jars on ice to prechill for at least 10 min. CAUTION: Methanol and acetone are toxic, wear gloves.
- Take a pair of bottom and top slides from the dry ice, swiftly twist them apart, and immediately immerse the "bottom" slide in ice cold methanol for 2 min. The "top" slide may be discarded.
- Transfer slides from methanol to ice cold acetone for 4 min. If the slides had many worms, most (90%) will fall off in the fix, even with the best prepared slides. If single worm slides were properly prepared, the worms should stay adhered.
- Place or dip the slides in the jar with PBS (to rinse off fixative) and proceed to step 4 (staining).
3B. Hard fix using formaldehyde or glutaraldehyde
- Dilute reagent grade formaldehyde or glutaraldehyde solution in 1x PBS to 0.5-4% final concentration. Aliquots (1.5 ml) may be stored tightly capped at -20 to -30 °C; defrost aliquots on ice just before using. NOTE: formaldehyde solutions degrade over time and with exposure to air. CAUTION: formaldehyde and glutaraldehyde are toxic, wear gloves and work in the hood.
- Take a pair of bottom and top slides from the dry ice, swiftly twist them apart, and immediately place the "bottom" slide with worms in the flat dish with lid. (The top slide can be discarded).
- Immediately pipette 200 μl toxic fixative over the center of the area of the slide with worms. The fixative will partially freeze in place, then melt to cover a larger region of the slide.
- If the worms are not totally covered with fixative, immediately and carefully add more fixative using a micropipette. Do not allow fixative to flow over the edge of the slide.
- Cover the dish with a lid and leave for 10 min to overnight at room temperature. Be sure the dish is humidified if longer incubations are used – this can be done by adding folded wet lint-free wipes to the dish. Do not let the wipes touch the slides.
- After fixation is complete, carefully pipette the fixative with loose worms into a 1.5 ml tube and dip the slide in a staining jar with PBS.
- Spin the tube with worms that came loose at high speed for 30 sec to pellet. Rinse the worms twice with PBS, then process in parallel with the worms on the slides (doing steps in microfuge tubes, rather than on slides).
- Proceed to step 4 (staining).
4. Antibody Staining
The antibody staining protocol is similar to standard protocols for any tissue on slides. This protocol is the same for all slides regardless of the preparation in steps 1-3.
- Prepare Antibody Buffer by mixing 700 ml double distilled water, 100 ml 10x PBS, 5 ml Triton X-100 (0.5% final), 2 ml 0.5 M EDTA pH 8 (1 mM final), 1 g BSA (0.1% final), 5 ml 10% sodium azide solution (0.05% final). pH to 7.2 with HCl, then add double distilled water to 1 L; store at 4 °C.
- Prepare Block by reconstituting donkey serum with double distilled water, then adding 5 ml serum to 45 ml antibody buffer (final 10% serum).
- Prepare primary and secondary Antibody Solutions by diluting antibody in antibody buffer plus 1% BSA. Recommended dilutions are 1:50-1:2,000 for primary antibodies and 1:1,000-1:5,000 for secondary antibodies (Cy3or OG488 conjugated donkey antibodies against species used to raise primary antibody).
- Prepare DAPI stock by mixing 2.5 mg/ml in double distilled water; store in 8 μl aliquots frozen at -30 °C. CAUTION: DAPI is a toxic DNA binding dye, wear gloves while weighing and dispensing into aliquots.
- Prepare 10 ml Mounting Medium by mixing 200 mg n-propyl gallate, 0.3 ml 1 M Tris pH 9, 2.7 ml double distilled water in a 15 ml conical tube. Heat at 65 °C for about 10 min until dissolved. Add 7 ml glycerol and an 8 μl aliquot of DAPI and vortex well. Aliquot medium into 1.5 ml tubes, wrap in foil, and store in dark at 4 °C (both air and light degrade the medium – if it becomes more yellow, discard in Biohazard waste). CAUTION: n-propyl gallate is a reactive anti-oxidant, wear gloves while weighing.
- Transfer slides to a staining jar with Block for 1 hr at room temperature or overnight at 4 °C (overnight preferable for hard fix). Avoid shaking to avoid losing worms.
- Transfer slides to staining jar with primary antibody or place slides flat in a humidified dish and top each with 100-500 μl primary antibody. Incubate slides overnight at 4 °C without shaking.
- After primary antibody incubation, transfer slides to staining jar of PBS.
- Filter Block through funnel lined with filter paper and store at 4 °C; if sterile filtered every 1-2 weeks, Block can be reused for 1 or more months. Similarly, filter and save primary antibody at 4 °C for reuse (for up to 10 or more rounds of staining).
- Rinse slides three times (~20 min each) in antibody buffer.
- Place slides in secondary antibody in a light-tight staining container and incubate 4 hr at room temperature or overnight at 4 °C.
- Transfer slides to staining jar of PBS. Filter and save secondary antibody at 4 °C for reuse (for up to 10 or more rounds of staining).
- Rinse slides three times (~20 min each) in antibody buffer.
- Transfer slides to PBS and leave in PBS (minutes to overnight at 4 °C) until ready to mount.
- Work rapidly on individual slides, so that the worms on front of slide stay wet. Remove one slide from the PBS, dry back of the slide with a lint-free wipe, and place on paper towel.
- Place ~20 μl of mounting medium over worms so that there are approximately equal parts medium and PBS on slide and tilt slide gently to mix the two solutions. CAUTION: Mounting medium is toxic.
- Tilt slide so frosted edge is lower and solution gathers near frosting, then place one edge of 24 x 60 mm coverslip on solution.
- Gently lower the coverslip to minimize air bubbles (which increase bleaching and disrupt viewing). If bubbles form, lift the edge of the coverslip slowly, then relower it without sliding the coverslip across the worms.
- Carefully dry off the edge of the slide without moving the coverslip, by compressing the edges with a lint-free wipe. The relatively dry edge of the slide must be visible all around the edge of the coverslip
- Seal the coverslip with 2-3 layers of nail polish. Slides can be placed in a flat slide holder during preparation and storage to protect from light. Once slide is well sealed and dry, clean the coverslip and back of the slide.
- Well-sealed slides can be kept in the dark for days at 4 °C or weeks at -20 °C. Over longer periods, DAPI staining of DNA and most green dyes (e.g. 488 nm excitation) fade. Reseal the coverslip with more nail polish as needed, e.g. after using any oil immersion lens.