Note: This protocol can be used to identify genes required for a variety of biological processes in a variety of tissue types. As a necessary quality control during the screen, animals are fed both positive and negative control dsRNA-expressing bacteria to demonstrate RNAi effects in the targeted tissue.
Other published feeding protocols grow dsRNA-expressing bacteria in the presence of either 50 μg/ml ampicillin or 25 μg/ml carbenicillin 8-10. We have found that bacteria grown under either of these conditions lose the dsRNA plasmid at high frequency. Indeed, we have found that even when bacteria are grown overnight in very high concentrations (up to 2 mg/ml) of ampicillin more than 50% of the bacteria no longer contain plasmid. While growth in carbenicillin generally resulted in higher retention of plasmid, 25 μg/ml was not sufficient to prevent plasmid loss. Instead, we found that carbenicillin concentrations of 500 μg/ml were required to block plasmid loss during growth in liquid cultures and 2 mg/ml carbenicillin was required to block plasmid loss when bacteria were grown on solid agar substrates.
Because plasmid retention is critical to the success of knockdown, only carbenicillin is used when dsRNA bacteria are cultured in this protocol. We have carefully optimized the concentration of carbenicillin used in the bacterial growth cultures described in this protocol to ensure that bacteria that do not contain plasmid do not grow in these cultures. However, as a quality control measure, plasmid loss is determined at several steps of the protocol. To ensure efficient knockdown, more than 80% of the bacteria at each of these steps must contain dsRNA plasmid. If more than 20% of the bacteria at any step of the protocol have lost the dsRNA plasmid, check the quality of the carbenicillin used. Carbenicillin should be prepared fresh the day it is used. Prepare fresh carbenicillin and repeat the culture.
1. How to Determine Plasmid Loss
- Remove a small sample of bacteria (as described at each relevant step) and add it to 450 μl sterile water.
- Use 100 μl of this diluted sample to create further 1:10, 1:100, and 1:1,000 serial dilutions using sterile water, mixing the solutions thoroughly between dilution.
- Spread 100 μl of each dilution onto LB and LB AMP plates and incubate these plates overnight at 37 °C.
- The next day, identify the LB plate that contains between 100 and 500 bacterial colonies. Count the number of colonies on this plate and on the corresponding LB AMP plate (containing the same dilution of bacteria).
- Determine plasmid loss from these colony counts. The percent of bacteria that do not contain the plasmid is calculated using Equation 1, and should be less than 15% for effective dsRNA knock down.
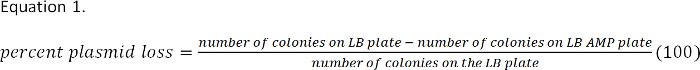
2. Making Duplicate Library Plates
Overview: The dsRNA library will arrive from the manufacturer (OpenBiosystems) as 10,566 bacterial clones in 96 well plates. Store the plates immediately in -80 °C freezer. The original library plates will be duplicated and all feeding screens will use the duplicate library plates as the source of bacteria. It is advisable to store the original and duplicate library plates in separate freezers. For ease of handling, duplicating only 10 library plates per day is recommended.
- Remove library plate(s) from -80 °C freezer and remove the foil seal while cultures are still frozen. Replace the library plate’s plastic cover and set the plate on a level surface to thaw at room temperature (no longer than 30 min).
Note: We have noticed that a significant percentage of bacteria found in each well of the original library plates do not contain plasmid. However, because the library bacteria are a precious resource it is not recommended to test for plasmid loss in the original library cultures.
- Using a multichannel pipette, add 150 μl of library duplication media to each well of an empty 96-well flat bottom duplicate plate.
- Mix the thawed library bacterial cultures by pipetting up and down four to five times using a multichannel pipette set to 100 μl. Then remove 10 μl of each mixed culture and transfer to the corresponding wells of duplicate plate. Continue to transfer cultures from library plate to duplicate plate making certain to change pipette tips for each transfer.
- After all cultures from a library plate have been transferred, cover the wells of the original library plate with new adhesive foil using a brayer roller to ensure a thorough seal. Replace the plastic lid on the plate and place it upright in a -80 °C freezer until the cultures have refrozen (at least 30 min). Then move the original library plates back into a library storage box.
- Shake duplicate library plate cultures flat for 8-10 hr at 37 °C (450 rpm on a microshaker, orbit 3 mm).
- Determine plasmid loss in each duplicate library plate. We found that a high proportion of the bacteria in each well of the original library plate did not contain dsRNA plasmid. It is important that these bacteria do not contribute to growth in the duplicate library. To prevent their growth, a high concentration of carbenicillin is used in the library duplication media (3 mg/ml). After culture of the duplication plate using these conditions we found that more than 90% of bacteria contain dsRNA plasmid. Remove 10 μl sample from a representative culture well of each duplicate plate and add it to 450 μl sterile water. Using these diluted samples, determine plasmid loss in each duplicate library plate according to steps 1.1-1.4. If significant plasmid loss is observed (>20% of bacteria in the duplicate library do not contain plasmid), remake the culture media using fresh carbenicillin and repeat protocol from step 2.1.
- After incubation, place a new foil adhesive sheet directly over the duplicate plate wells, and place the plate lids over this foil. Place the duplicate library plates upright in a -80 °C freezer (at least 30 min) until frozen. Once frozen, move the plates to the duplicate library storage box for long-term storage.
3. Making Temporary Copies of the Library on Omniplates
Overview: To begin to prepare food for a dsRNA screen, bacteria from duplicate library plates are transferred to solid agar omniplates and grown overnight. To prevent plasmid loss on solid media, omniplates contain carbenicillin at 2 mg/ml. The bacteria on these plates can be used for a period of one week, and show no plasmid loss. We have not tested for plasmid loss in bacteria grown on omniplates longer than one week.
- Remove duplicate library plate(s) from -80 °C freezer and take off the foil seal while the cultures are still frozen. After removing the foil, replace the plate’s plastic cover and set the duplicate library plate on a level surface to thaw at room temperature (no longer than 30 min).
- Sterilize the Boekel 96 pin replicator. The pins of a clean replicator should be rinsed with distilled water and then sterilized before each use. To sterilize the replicator, place it into an ethanol bath (3/4 in deep in a Pyrex dish) and then burn the ethanol coating the pins off using a Bunsen burner. Allow the flame to extinguish and then repeat.
- Place the sterilized replicator into the wells of the thawed duplicate plate and use it to gently stir the cultures. Small volumes of the culture will adhere to the pins of the replicator.
- Carefully remove the replicator from the wells of the duplicate plate and place it (pins down) gently on the surface of the omniplate, being careful not to pierce the agar surface. Leave the replicator on the surface of the omniplate for 3-4 sec so that bacteria from the pins of the replicator are transferred onto the agar surface.
- Remove the replicator and allow the small volume (2-3 μl) of bacterial culture to absorb into the agar. Incubate the omniplates inverted for 15-18 hr at 37 °C. These omniplates can be used the next morning to inoculate 96 well liquid cultures. Alternatively, omniplates containing overnight bacterial colonies can be stored up to seven days at 4 °C.
- To replica-transfer additional duplicate plates, rinse the replicator tips with sterile water and repeat the sterilization process by placing the replicator in ethanol and flaming it twice, as before. Once sterilized, the replicator can be used on the next duplicate plate.
- Determine plasmid loss after overnight culture on omniplates. The bacterial colonies on the omniplates after overnight growth will serve as a temporary source of food for feeding screens. It is important that more than 80% of the bacteria in each colony contain dsRNA. Bacteria tend to lose plasmid more quickly on solid agar media than in liquid cultures. Determine plasmid loss for two representative colonies on the omniplates by inoculating 1 ml of sterile water with a small amount of bacteria from each colony and using this diluted sample to determine plasmid loss according to steps 1.1-1.4. Expectation: More than 90% of bacteria in each colony will contain dsRNA plasmid. If significant plasmid loss is observed (>20% of bacteria from a colony on the omniplate do not contain plasmid), remake the omniplate media using fresh carbenicillin and repeat protocol from step 3.1.
4. Preparation of Bacteria for Feeding Worms
Overview: Bacteria for feeding worms are prepared as 1 ml liquid cultures (in a 96 well format) using bacteria from the omniplates. These liquid cultures are grown overnight to generate saturated nonlogarithmic bacterial cultures. Overnight growth ensures that all cultures will contain similar concentrations of bacteria and therefore all worms will be fed the same amount of food. No plasmid loss during this growth should be observed.
- Dispense 1 ml of bacterial growth media into each well of 2 ml deep-well 96-well plate using a repeat pipettor and 50 ml Combitip.
- Place sterile replicator onto the surface of omniplates containing bacterial colonies generated in steps 3.1-3.6 making certain that pins are in contact with each of the 96 bacterial colonies.
- Carefully move the replicator from the surface of the omniplate into the deep well plate making certain that the pins do not scrape the sides of the deep wells until immersed in growth media. Move the replicator slowly in a square motion following the inside wall of the deep well plate to dislodge the bacteria into the growth media. Be careful not to splash and cross contaminate wells.
- Control wells: For each 96-well deep well culture plate add a dsRNA-expressing bacteria to one well that will act as a positive control for the expected knockdown phenotype and add bacteria containing empty dsRNA vector (pL4440) as a negative control to another well. We re-streak control dsRNA-expressing bacteria once per week onto LB plates containing tetracycline (15 μg/ml) and carbenicillin (2 mg/ml) so that the bacteria remain fresh and retain plasmid.
- Using a sterile inoculating loop remove a single well-isolated colony from the control plate (about the same quantity of bacteria transferred by the replicator) and inoculate an empty well in the deep well culture plate. Record the position of the well that has been inoculated. Alternatively, the loop with bacteria can be moved to an Eppendorf tube containing 250 μl growth media, vortexed and then used to inoculate several wells.
- Shake the deep-well plates in a flat position at 650 rpm on microshaker (orbit: 3 mm) at 37 °C overnight. The cultures will be saturated by the next morning. Regardless, the concentration of carbenicillin used in these cultures should prevent plasmid loss.
- Determine plasmid loss in the 1 ml overnight cultures. The cultures will be used directly to feed worms for the screen. It is important that more than 80% of the bacteria in each culture contain dsRNA plasmid. Determine plasmid loss for two representative cultures according to steps 1.1-1.4. Expectation: More than 90% of bacteria in each culture will contain dsRNA plasmid. We have never observed significant plasmid loss in these overnight cultures even when grown to saturation. However, if significant plasmid loss is observed (>20%), remake the growth media using fresh carbenicillin and repeat protocol from step 4.1. Cultures can be restarted from the original omniplates as long as the omniplates are less than 1-2 weeks old.
- Once used to start 1 ml cultures, the omniplates are stored at 4 °C until the screen is completed and can be used as a source of food to start any retest cultures.
- After overnight incubation of cultures, use a 12-channel multichannel pipette to remove 150 μl of bacterial culture from the deep well plate and eject onto the surface of the feeding plate. It is important not to pierce the surface of the agar during transfer as worms will burrow and not feed on the dsRNA-expressing bacteria. Transfer can be done four wells at a time (Figure 1). To do this, place pipette tips on every third channel of the multichannel pipette (as there are only 4 wells per row in the feeding plate). After each set of 4 wells is transferred, change to new, sterile pipette tips. Each 96 deep-well culture plate will require 96 tips for transfer to eight 12-well feeding plates.
- Store seeded plates upright in the dark overnight so bacteria can absorb into the agar.
5. Generating L1 Arrested C. elegans Larvae
Overview: L1-arrested larvae are used to start synchronized worm cultures on the dsRNA feeding plates. These larvae are generated by bleaching populations of gravid adults to isolate healthy eggs and then allowing the eggs to hatch in S-complete media without food. In the absence of food the hatched L1 larvae will arrest development, generating a synchronous population of L1 larvae. During the screen, prepare fresh L1-arrested larvae every week as these starved animals become sick over time and must be discarded. The choice of genetic background of the animals used in a dsRNA feeding strain can vary depending on application, for neuronal screens eri-1; lin-15B mutants are recommended.
- Pick 10-20 L4 larvae to six separate NGM plates containing a lawn of OP50 bacteria and wait 6-7 days until the plates contains many freshly-laid eggs and gravid adults.
- Remove animals and eggs from the plates by adding 3 ml of water to the surface of each plate and use a gloved finger to dislodge the eggs, bacteria, and animals from the surface of each plate into the water. Be sure to cover all areas of the plate, paying special attention to the bacterial lawn. Remove the water, bacteria, worms and eggs from each plate using a glass pipette and transfer to one sterile 50 ml conical tube.
- Add another 3 ml of water to the surface of each plate, pipetting up and down across the surface to remove all remaining bacteria, worms and eggs. Add this volume to the 50 ml conical tube.
- Spin the conical tube at 1,500 rpm for 1 min in a table top centrifuge. Worms and eggs should pellet to the bottom of the tube and the bacteria should remain suspended. Carefully aspirate all but 4 ml of the liquid from the tube being sure to avoid the pelleted worms and eggs.
- Resuspend the worm pellet in the residual water and transfer to a sterile 15 ml conical tube using a glass pipette. Bring the volume to 10 ml with sterile water.
- Spin at 1,500 rpm for 1 min as before. Aspirate the supernatant leaving 200 μl water so as not to disturb the worm/egg pellet.
- Add 10 ml water to the conical to wash the worms and eggs and spin as before to remove additional bacteria.
note: In steps 5.8-5.11 worms and eggs are exposed to a bleach solution. Bleach will destroy worms and leave eggs relatively unharmed. However, if eggs are left in bleach solution too long, they will also be destroyed. Set a timer when bleach is added in step 5.8 to be certain that eggs do not remain exposed to bleach for more than 10 min.
- Remove all but 200 μl of the supernatant, again avoiding the pellet, then add 10 ml bleach solution and start a timer.
- Incubate worms and eggs in bleach solution for 3-4 min, occasionally mixing by inversion. Then spin at 1,500 rpm for 45 sec in a tabletop centrifuge. Look for a pellet in the bottom of the tube, it should be smaller and more compact than the original pellet and might take on a yellow appearance.
- Aspirate the bleach solution leaving 200 μl behind so as not to disturb the pellet.
- Add another 10 ml fresh bleach solution and invert as before. When the timer reads 10 min, spin again and aspirate the bleach solution, leaving 200 μl behind and quickly add 10 ml sterile water. Examine the solution under a dissecting microscope. The larvae and many of the adults should have dissolved in the bleach solution, leaving mostly eggs behind.
- Spin as before for 45 sec, and again aspirate the supernatant so as not to disturb the pellet. This pellet should contain primarily eggs and will be very loose.
- Add another 10 ml water, invert to mix, spin and aspirate leaving a little water behind. It is important to remove as much bleach solution as possible.
- Add 4 ml S-complete media to the conical tube, resuspend the eggs and transfer to a sterile 100 ml Erlenmeyer flask. Place the flask in a 20 °C incubator on a shaker set moving just fast enough to cause the contents of the flask to swirl. This will provide sufficient aeration to support the L1 larvae when they hatch. Within 15 hr all of the eggs should have hatched producing a synchronous population of L1 arrested larvae.
Arrested L1 larvae should be made fresh every week during a screen. Once the first batch has been made, subsequent batches can be started by adding 500 arrested L1 larvae (from the previous week’s batch) to each of four standard, seeded 6 cm plates (containing 100 μl overnight culture of OP50), and incubated at 20 °C for 4 days. On the fourth day the plates should contain many eggs and gravid adults and can be bleached to prepare fresh eggs for more synchronized L1 larvae.
6. Transferring Arrested L1 Larvae onto Feeding Plates
Overview: To perform a dsRNA screen, 20-30 L1 arrested larvae are transferred onto dsRNA feeding plates. Early knockdown effects such as larval arrest or lethality will be observed after animals have fed on dsRNA-expressing bacteria for 2-3 days. Depending on the screen conducted, desired phenotypes may be observed either in the original animals placed on the feeding plate (P0 animals) or in their progeny (F1 animals). The presentation of phenotype will depend on a number of variables including the perdurance of the protein encoded by the gene whose expression is knocked down and the time during development in which the gene of interest is required. Starting dsRNA feeding cultures with only 20-30 animals should permit observation of both P0 and F1 phenotypes before animals exhaust the food source.
To easily transfer 20-30 animals to feeding plates, first determine the concentration of L1 arrested animals in the arrested L1 culture.
- Mix the Erlenmeyer flask containing the arrested L1 larvae to distribute the animals.
- Remove a 10 μl aliquot and place drop-wise onto an unseeded 6 cm NGM plate in 4-5 drops down the center of the plate making sure that all media is expelled from the pipette.
- Using the end of the pipette tip spread the dots of worm suspension to form a single continuous line of liquid that spans the diameter of the plate.
- Using a dissecting microscope, count all animals starting at one end of the line of liquid and continuing to the other end before the liquid has absorbed into the plate and the animals disperse. This may require constant refocusing of the dissecting scope to be certain that no animals are missed.
- Repeat this for three separate 10 μl aliquots and average the three numbers to determine the average number of worms per μl of worm suspension and record this number directly on the Erlenmeyer flask.
7. Begin the Feeding Screen
Overview: To begin the dsRNA screen, 20-30 arrested L1 larvae are added to each well of 12-well feeding plates containing a thin lawn of dsRNA-expressing bacteria. Feeding plates contain standard nematode growth media (NGM) and are supplemented with 500 μg/ml carbenicillin (to prevent plasmid loss) and 1 mM IPTG (to induce dsRNA expression). Animals are allowed to feed and develop for several days at 20 °C. Two typical feeding regimens for animals are 3 days when performing a P0 screen and 6-7 days when performing an F1 screen. The duration of feeding, however, can be varied depending on the specific phenotypes sought in the screen.
- Use the suspension of L1 arrested larvae and sterile water to prepare a solution containing 2,000 worms/ml. Each 12-well feeding plate will require 120 μl of this worm suspension. Mix thoroughly to ensure an even distribution of animals and transfer the animals to a sterile reservoir for pipetting.
- Using the multichannel pipette set up with tips in every third position (see Figure 1) transfer 10 μl of the worm solution directly onto the bacterial lawn of the feeding plates. Take care not to pierce the surface of the agar, as worms will burrow into the agar and not feed on the dsRNA-expressing bacteria.
- Remix the worm solution in the reservoir (by gently sloshing it about) after every two rows of feeding plates, as worms settle quickly. Continue dispensing the worm suspension until all feeding plates have worms. Examine a few wells early on under the dissecting scope to ensure that each well is getting 20-30 worms.
- Store plates upright in a humidified box in a 20 °C incubator. The next day, after the worm suspension has absorbed into the plate, invert the plates and return to the humidor at 20 °C. Animals on these plates can be observed after 3 days (for P0 phenotypes) and again after 6 days (for F1 phenotypes).
note: The bacteria that remain on the feeding plate after 7 days of worm growth have been tested for plasmid loss and nearly 100% retained the dsRNA plasmid.
P0 knockdown phenotypes will begin to appear after 2-3 days of feeding animals dsRNA expressing bacteria. Some early phenotypes include larval arrest and lethality and should be observed in 2-3% of all feeding wells. These same phenotypes will also be observed in the F1 generation animals. The presence of F1 eggs that fail to hatch is another common F1 phenotype, and together, these three phenotypes will be observed in nearly 10% of all wells in F1 screens.
We used the protocol described here to examine knockdown effects for several genes expressed in a variety of tissue types for both P0 and F1 phenotypes. Because we also wanted to test for knockdown effects in neuronal tissue, we used eri-1; lin-15B mutants in our screens. Mutations in eri-1 and lin-15B, along with mutations in rrf-3, were identified in screens for mutations that caused enhanced sensitivity to RNAi 8,13,14.
We tested four genes for knockdown: egl-30, dpy-17, pat-10, and unc-4. Two of these genes, egl-30 and unc-4, are expressed in the nervous system 15-17, one gene, pat-10, is expressed in body-wall muscle 18, and the fourth gene, dpy-17, is expressed in hypodermal cells 19. When we fed eri-1; lin-15B animals bacteria that contained the empty pL4440 plasmid but did not express a dsRNA, we did not observe any morphological or behavioral defects (Figure 2A, Table 1).
egl-30, encodes the C. elegans ortholog of the G protein subunit Gaq. egl-30 null mutants arrest during early larval development, but some null escapers and hypomorphic loss-of-function egl-30 mutants grow to become adult stage and are defective in locomotion and egg-laying behaviors 20. When we fed L1 stage eri-1; lin15B larvae bacteria expressing egl-30 dsRNA, we found that the larvae grew to become adults that showed clear defects in locomotion and egg-laying behavior. After three days, 100% of the animals fed dsRNA against egl-30 were paralyzed and unable to lay eggs (Figures 2B, Table 1). We did not observe the larval arrest phenotype in our dsRNA knockdown experiments that is observed in egl-30(ad810) null animals. This is presumably because the L1 animals we plated onto egl-30 dsRNA bacteria had already passed the early developmental requirement for EGL-30. This highlights an advantage of feeding screens: late-stage phenotypes can be observed for genes that also play vital roles during development.
dpy-17 encodes a collagen protein required for cuticle formation in C. elegans 19. dpy-17 null mutants are shorter and fatter than wild-type animals. We did not observe any morphological defects in P0 animals fed dpy-17 dsRNA. However, 98% of the F1 animals showed the Dpy phenotype (Figure 2C, Table 1). The lack of short and fat P0 animals indicates that dpy-17 gene function is required at early larval stages for proper body morphology. While dpy-17 has not been targeted in other feeding screens, it was knocked down by dsRNA injection 21. Interestingly, only 30% of the progeny of dsRNA injected animals showed the Dpy phenotype, suggesting that the feeding protocol described here is more efficient than the more labor-intensive injection approach, at least for some genes.
pat-10 encodes body wall muscle troponin C, a protein containing four EF Hand motifs that bind calcium 18. We found that 100% of eri-1; lin-15B animals fed pat-10 dsRNA became paralyzed within 3 days and laid no eggs leading to a lethal phenotype (Figure 2D, Table 1).
unc-4 encodes a homeodomain protein expressed in ventral cord motor neurons and is required for proper synaptic input choice 22,23. We chose unc-4 as a test gene because it has proven to be resistant to previous dsRNA feeding strategies 8,24. unc-4 mutants show a defect in locomotion behavior. As a result of defects in synaptic connectivity, unc-4 mutants are unable to back 22,23. Compared to wild-type animals that back freely in a sinusoidal motion when prodded on the head, unc-4 null mutants do not back and instead contract their midbody tightly, causing a dorsal flexure which often becomes so extreme that the head and tail of the animal touch, placing the body in a coiled position. We found that 68% of animals fed unc-4 dsRNA-expressing bacteria from our library preparation showed defects in backing. This is a dramatic improvement in knockdown efficiency when compared to the 0% backing defect observed when rrf-3 animals are fed unc-4 dsRNA using previously published protocols 8 (Table 1).
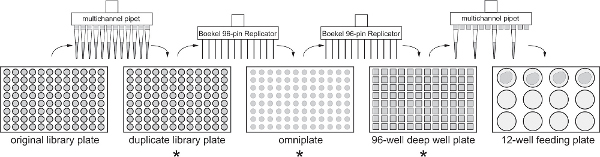
Figure 1. Flow chart for library duplication and large scale screen. Each 96 or 12 well plate generated during the screening protocol is indicated, along with the method used to transfer bacteria between plates. Asterisks indicate the steps at which bacteria should be tested for plasmid loss. Click here to view larger figure.
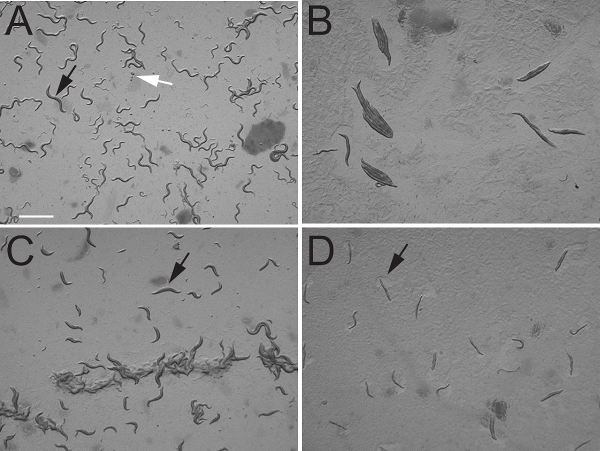
Figure 2. dsRNA knockdown phenotypes of C. elegans genes expressed in various tissue types. A) Control animals fed bacteria containing pL4440 empty vector. Black arrow indicates the position of a young adult animal. White arrow indicates the position of a group of laid eggs. Photograph shows freely moving animals as evidenced by their normal body posture. B) Animals fed dsRNA against egl-30. Note that all animals have adopted a rigid appearance typical of paralyzed animals. Also note the complete absence of laid eggs on the plate. C) Animals fed dsRNA against dpy-17. Note that adult animals in the field (one marked by arrow) are shorter and fatter than the adult animal shown in panel A. D) Animals fed dsRNA against pat-10. Note that all animals are paralyzed but can still move their head muscles to feed as indicated by the clearing of bacteria near the head, marked by arrow. Scale bar 1 mm.
| dsRNA plasmid or gene targeted by feeding | Tissue expression of targeted gene | Percent animals with terminal phenotype |
| pL4440 (negative control) | wildtype for all behaviors | |
| egl-30 | nervous system | 100% Egl and Paralyzed |
| dpy-17 | hypodermis | 98% Dpy |
| pat-10 | body wall muscle | 100% Paralyzed |
| unc-4 | nervous system | 68% Backing Defect |
Table 1. Terminal phenotype of animals fed dsRNA against genes expressed in various C. elegans tissues. n = 100 for all genes.
