1. Preparation of bacterial cultures.
- Grow all cultures in Erlenmeyer flasks with a culture/flask volume ratio of at most 1/6. In a typical experimental setup, grow the cultures at 37.0 °C and shaking at 160 rpm in morpholinepropanesulfonic acid (MOPS) minimal medium18 supplemented with the preferred carbon source, for example 0.2% glucose, for at least 10 consecutive generations before RNA harvest.
- The day before RNA harvest, dilute an outgrown culture of the desired strain in MOPS minimal medium in such a way that it will reach the desired OD436 at the desired time the following day. Calculate the volume of outgrown culture to use for inoculation by the formula:
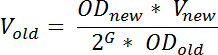
NOTE: Here, ODnew denotes the desired OD436 after incubation, Vnew denotes the volume of the diluted culture, ODold denotes the OD436 of the outgrown culture diluted from, G denotes the number of generations the culture will grow, from the time of dilution (t1) to the time it is needed the day after (t2) and can be calculated as: time difference (in min) between t1 and t2 divided by the generation time (in min). To ensure steady-state growth at the time of sampling, G should be ≥10; Vold denotes the volume of the out-grown culture that should be transferred to the fresh medium. This equation is an approximation that does not account for any lag phase; a phenomenon often observed after dilution of an out-grown culture in fresh media.- Growth conditions will vary depending on the purpose of the experiment, but to limit cell-to-cell variation in tRNA levels and to increase the reproducibility of the results, (recommended) grow the cultures to steady state prior to sampling19.
2. Preparation of spike-in cells
NOTE: The E. coli strain MAS1074, which expresses the selC gene encoding tRNAselC under the control of an isopropyl β-D-1-thiogalactopyranoside (IPTG)-inducible promoter is used as the spike-in strain.
- Dilute an outgrown culture of MAS1074 to an OD436 of 0.05 in MOPS minimal medium supplemented with 100 µg/mL ampicillin and 0.2% glucose. Grow the culture at 37 °C shaking at 160 rpm.
NOTE: See the Discussion for the use of an alternative reference strain. - At OD436 ~0.1, add IPTG to a final concentration of 1 mM to induce the expression of tRNAselC. Grow the culture to OD436 ~0.5 (~3-4 h of growth) and move the culture flask to an ice bath. Keep the spike-in cell culture on ice until all samples for RNA purification have been harvested.
3. Harvest of experimental samples.
- Pre-warm (37 °C) one capped 100 mL Erlenmeyer flask (or similar) containing 4 mL 10 % (w/v) trichloroacetic acid (TCA) for each sample by placing it in a heated water bath.
NOTE: CAUTION, TCA decomposes upon heating, producing toxic and corrosive fumes including hydrogen chloride and chloroform. The solution in water is a strong acid; it reacts violently with bases and is corrosive to many metals. Proper precaution should be taken when using this liquid. - Immediately prior to cell harvest, measure and record the OD436 of the culture.
- To harvest the cells, transfer 4 mL of culture to a pre-warmed flask containing TCA.
- Mix the sample by swirling the flask by hand for 5 s. Mixing with TCA immediately disables all enzymatic activities and thus preserves the charging levels of the tRNAs.
NOTE: If multiple samples are to be collected, keep harvested samples in TCA on ice until all samples have been collected.
4. Addition of spike-in cells
NOTE: Preparation of spike-in cells is described in Step 2.
- When all samples for RNA preparation have been collected, add 5% spike-in cells (based on OD) to each sample.
NOTE: The volume of spike-in cell culture needed is calculated by the formula:
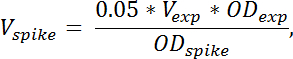
where Vspike denotes the volume of spike-in cell culture needed, Vexp denotes the volume of harvested experimental cell culture (without TCA), ODexp denotes the OD436 of the experimental cell culture at the time of harvest into TCA. ODspike denotes the OD436 of the spike-in cell culture. If multiple samples are harvested at different ODs, the volume of spike-in cells added should be adjusted according to the formula above for each sample. - To quantify the contribution from the spike-in cells to the RNA species of interest, prepare a sample only containing spike-in cells by mixing 4 mL spike-in cells with 4 mL TCA. Prepare RNA from this sample in the same way as for the other samples.
- Optionally, take another flask containing only the experimental sample, without spike-in cells, and include it to quantify the contribution of the experimental sample to the tRNAselC levels; the endogenous levels of tRNAselC di E. coli K-12 are negligibly low.
5. Preparation of RNA
NOTE: The RNA preparation protocol described here is essentially that described by Varshney et al.1; To avoid deacylation of the aminoacylated tRNAs during purification it is important to keep the samples at acidic pH and at 0 °C throughout the course of purification. Use clean sterile tubes/flasks to hold the samples and solutions, and prepare all solutions with ultrapure (18.2 MΩ·cm resistivity) water.
- Pour 8 mL of each sample into an ice-chilled centrifuge tube. Centrifuge samples for 10 min at 9,500 x g at 4 °C. Completely remove supernatant and resuspend in 0.3 mL cold 0.3 M sodium acetate, pH 4.5 and 10 mM EDTA.
- Transfer each sample to a cold 1.5 mL microcentrifuge tube and add 0.3 mL phenol equilibrated with the same buffer.
NOTE: CAUTION, phenol produces toxic fumes and is highly corrosive to the skin. - Vortex each sample 10x for 15 s with at least 1 min between each vortex. In between vortexing keep the samples at 0 °C. Use the frequent pauses to ensure that the samples remain cold.
- Centrifuge samples for 15 min at 20,000 x g at 4 °C. Transfer the water phase (upper phase) to new 1.5 mL microcentrifuge tubes. Add 0.3 mL cold phenol and vortex 4x 15 s as before.
- Centrifuge for 10 min at 20,000 x g at 4 °C. Transfer the water phase to new, cold 1.5 mL microcentrifuge tubes containing 2.5 times the volume of 96% ethanol (~750 µL). Precipitate the RNA by incubating the tubes for 1 h at -20 °C or overnight at -80 °C.
- Centrifuge samples for 30 min at 20,000 x g at 4 °C. Discard the supernatant and carefully add 1 mL cold 70% ethanol without disturbing the RNA pellet. Gently invert once to wash the insides of the tubes with ethanol.
NOTE: The pellet can be hard to see at this step. - Centrifuge for 10 min at 20,000 x g at 4 °C. Completely remove the supernatant and air-dry the pellet until no ethanol is left. Resuspend the pellet by vigorously vortexing in 20 µL cold 10 mM sodium acetate pH 4.5 and 1 mM EDTA.
NOTE: Do not over-dry the pellet as it will become difficult to resuspend. - Optionally, assess the concentration of RNA by measuring absorbance at 260 nm.
- Optionally, assess the quality of the purified RNA by agarose gel electrophoresis20.
- Store samples at -80 °C.
NOTE: The protocol can be paused here.
6. Preparation of chemically deacylated control sample
NOTE: To distinguish the band of aminoacylated tRNA from its deacylated counterpart on the Northern blot, a chemically deacylated aliquot is prepared by alkaline treatment. For most purposes, it is sufficient to deacylate a single sample for each Northern blot.
- Thaw the RNA sample on ice. Transfer a 4 µL aliquot of the RNA sample to a new tube. Add 46 µL Tris-HCl pH 9.0. Incubate for 2 h at 37 °C.
- Add 15 µL of 0.3 M sodium acetate at pH 4.5 and subsequently add 125 µL of 96% ethanol. Precipitate RNA at -20 °C for 1 h.
- Centrifuge for 30 min at 20,000 x g at 4 °C. Completely remove supernatant and resuspend in 4 µL cold 10 mM sodium acetate pH 4.5 and 1 mM EDTA.
NOTE: The protocol can be paused here, if the sample is stored at -80 °C.
7. Gel electrophoresis and Northern blotting
- Mix 4 µL sample (untreated or chemically deacylated) with 6 µL loading buffer (0.1 M sodium acetate (pH 5.0), 8 M urea, 0.05% bromophenol blue, and 0.05% xylene cyanol).
NOTE: Remember to include the sample containing only spike-in cells, the chemically deacylated sample (and the sample without spike-in cells, if one was prepared). - Load the samples onto a 0.4 mm thick 6.5% polyacrylamide gel (19:1 acrylam- ide:bisacrylamide) containing 8 M urea in 0.1 M sodium acetate buffer (pH 5.0). Use 60 cm long gels for proper separation of tRNA species.
- Separate the RNA by electrophoresis at 10 V/cm gel at 4 °C, until the bromophenol blue reaches the bottom of the gel (~20 h).
- The area from the xylene cyanol dye and 20 cm down towards the bromophenol blue dye is used for blotting (the distance between the two dyes should be 25-30 cm). Carefully separate the two glass plates, so that the gel remains on one of them. A thin piece of filter paper is cut to the size of the desired blotting area and placed on top of the part of the gel that will be used for blotting. The parts of the gel that are not covered by filter paper are cut off and discarded. Use the filter paper to carefully lift the gel piece away from the glass plate.
NOTE: The large gel is very fragile so handle with care. - A positively charged nylon membrane (e.g. Hybond N+ membrane) is placed on top of the gel and it is electroblotted at 20 V for 90 min using 40 mM Tris-acetate (pH 8.1), 2 mM EDTA as transfer buffer.
- Crosslink the RNA to the membrane using UV-light (0.12 J/cm2).
NOTE: The membrane can now be stored at room temperature and the protocol can be paused here.
8. Design and verification of probes
NOTE: Careful design and verification of Northern blot probes is crucial to obtain reliable results and avoid unwanted cross-hybridization with other RNA species.
- Use short synthetic oligonucleotides as probes. Design the probe sequence in a way that it is complementary to the most unique part of the sequence of the tRNA of interest – this is often the anticodon loop.
NOTE: A list of predicted tRNA sequences from various organisms can be found in the Genomic tRNA Database (GtRNAdb)21. - Adjust the length of the sequence to obtain a predicted melting temperature of 55-65 °C.
- BLAST (Basic Local Alignment Search Tool) the chosen sequence against the genome sequence of the bacterial strain used in the experiment, to verify that the sequence is unique to the selected tRNA, as described in22.
NOTE: Table 1 lists suggested probe sequences for several different tRNAs of E. coli K12. If the probe is specific for the tRNA of interest, only two bands are visible on the Northern blot (charged and uncharged tRNA) and only one band is visible in the lane with the deacylated sample (uncharged tRNA). If more than two bands are present or if further validation of probe specificity is desired, see Discussion.
9. Hybridization of Northern blot
NOTE: In the following steps, the membrane is sequentially hybridized with probes complementary to the desired specific tRNAs, including a probe specific for the reference tRNAselC.
- Place the membrane in a hybridization tube and add 6 mL hybridization solution (0.9 M NaCl, 0.05 M NaH2PO4 (pH 7.7), 5 mM EDTA, 0.5% (w/v) SDS, 100 mg/mL of sheared and denatured herring sperm DNA and 5x Denhardt's solution (100x Denhardt's solution = 2% bovine serum albumin, 2% polyvinylpyrrolidone 40, and 2% Ficoll)).
- Pre-hybridize the membrane in the buffer for 1 h rotating at 42 °C. Add 30 pmol radioactive oligo-DNA probe 5'-end-labeled with 32P (prepared as described by Sambrook et al.23)
NOTE: CAUTION, the high-energy beta emissions from 32P can present a substantial skin and eye dose hazard. Proper precaution should be taken when using 32P. Radioactive waste should be disposed of according to the appropriate institutional safety protocols. - Incubate the probe with the membrane overnight rotating at 42 °C. Remove the probe using a disposable 10 mL pipette.
NOTE: If stored in a freezer the probe can be reused. - In the hybridization tube, wash the membrane shortly in 50 mL of 0.3 M NaCl, 30 mM sodium citrate, 0.1% (w/v) SDS at room temperature.
NOTE: This first wash contains much unbound probe and must be handled as highly radioactive. - Repeat step 9.4 once, then move the membrane to a flat plastic container or tray including a lid. Cover the membrane with the washing solution (0.3 M NaCl, 30 mM sodium citrate, 0.1% (w/v) SDS) and incubate it for 30 min at room temperature.
- Change the washing solution and continue the washing until a satisfying signal to noise ratio has been obtained (often 3 – 4 changes of washing solution). Use a Geiger-Müller tube to detect how the noise to signal ratio changes by measuring the radiation from an empty area of the membrane and compare it to the area where the probe has hybridized specifically to tRNA.
- In order to be able to strip the probe from the membrane after exposure, ensure that (important) the membrane does not dry out before the stripping procedure. Place the membrane in a thin plastic bag or wrap it tightly using plastic wrap and seal it to be air-tight by tape or welding. Place the sealed membrane on a phosphorimaging screen.
NOTE: The required exposure time on the phosphorimaging screen is determined by the specific activity of the probe, the amount of RNA probed for, and the hybridization efficiency of the probe, and can vary greatly; from a few minutes to several days. With experience, the intensity of the radiation detected on the membrane with the Geiger-Müller tube gives a good indication of the required exposure time. For reliable quantification, it is important that the screen is not overexposed. - Scan the phosphorimaging screen using a laser scanner and save the file in ".gel" format.
NOTE: A high signal to noise ratio is necessary for reliable quantification. Probing for a tRNA should result in two bands; one containing aminoacylated tRNA (slower migration) and one containing deacylated tRNA (faster migration). See Figure 2.
10. Stripping the membrane
NOTE: Before the membrane can be probed for another tRNA, the previous probe must first be removed by stripping the membrane.
- Heat enough stripping buffer (15 mM NaCl, 1.5 mM sodium citrate, 0.1% (w/v) SDS) to cover the membrane and pour it over the membrane. Incubate it for 15 min at room temperature.
- Repeat step 10.1 until no radiation can be detected with a Geiger-Müller tube; the membrane can now be re-hybridized following the steps described in Section 9.
11. Quantifying tRNA and charging levels
- Load the .gel file into the imaging software (see the Table of Materials). Draw a vertical line along each of the sample lanes in such a way that it spans most of the width of the lane but excludes the edges of the bands where the signal can be uneven, as shown in Figure 3A.
- Visualize counts from each lane by clicking "Analysis" -> "Create Graph". Manually define the two peaks representing the charged and the uncharged tRNA from each lane, as shown in Figure 3B.
- Obtain counts from each peak by clicking "Analysis" -> "Area Report", and record the area under each of the two peaks.
- When the blot has been probed for tRNAselC and at least one tRNA of interest, normalize the levels of the tRNA of interest to tRNAselC.
- Calculate the proportion of tRNA originating from the spike-in cells in each lane and subtract from the total signal in that lane using the equation:
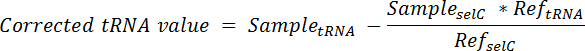
NOTE: Here, SampletRNA = the signal obtained from a single lane of a membrane probed for the desired tRNA; SampleselC = the signal obtained from the same lane of the same membrane probed for tRNAselC; ReftRNA = the signal obtained from the lane containing only spike-in cell RNA of the same membrane probed for the desired tRNA; RefselC = the signal obtained from the lane containing only spike-in cell RNA of the same membrane probed for tRNAselC. - To normalize, divide the corrected tRNA value from each lane by the tRNAselC signal from the same lane.
NOTE: The contribution to the tRNAselC signal from the sampled cells is negligible (See Figure 2, lane labeled "-selC").
- Calculate the proportion of tRNA originating from the spike-in cells in each lane and subtract from the total signal in that lane using the equation:
- Calculate the tRNA charging level for each lane by dividing the signal of the peak representing charged tRNA with the sum of the signal from both peaks (charged + uncharged).
Using the procedure described here, the abundance and charging levels of three tRNAs were measured in E. coli K-12 before and during amino acid starvation.
The arginine auxotroph strain NF9154 (thr leu his argH thi mtl supE44) was grown for at least ten generations in MOPS minimal medium supplemented with 0.4% glycerol, 50 µg/mL threonine, leucine, arginine, and 5 µg/mL histidine at 37 °C shaking at 160 rpm. Arginine starvation was introduced by filtration of the culture and resuspension in fresh MOPS medium as above but without arginine. During arginine starvation, protein synthesis is strongly reduced, but continues at a low level using arginine released by the turnover of existing proteins. Samples were harvested at the time points indicated in Figure 4. After all samples were harvested into TCA, 5% of the MAS1074 spike-in cells overexpressing tRNAselC, was added to each sample as illustrated in Figure 1. RNA was purified from the samples, separated by electrophoresis, and blotted onto a membrane as described in the protocol. The membrane was probed for tRNAargVYZQ, tRNAgltTUVW, tRNAhisR, and tRNAselC, as seen in Figure 2. The radiation signal from each lane was quantified for each of the four probes as illustrated in Figure 3, and corrected and normalized as described in Section 11. The results are presented in Figure 4. Figure 4A shows that the levels of all the tested tRNA species rapidly decrease during amino acid starvation, as recently reported4. Figure 4B shows that the tRNA charging levels behave as expected: the charged fraction of the arginine-accepting tRNA drops rapidly upon removal of arginine from the growth medium, whereas the charging levels of the other tRNAs increase slightly upon starvation because charged tRNAs accumulate when their rate of decharging at the ribosomes decreases due to the lack of substrate for protein synthesis4. Further into the starvation period, the charged tRNA fraction decreases to approximately pre-starvation levels due to the degradation of the majority of the tRNA pool (Figure 4A), which results in a greater rate of decharging of the remaining tRNAs at the ribosomes. By contrast, the charged fraction of tRNAargVYZQ increases for at least two reasons; 1) activation of the stringent response means that mRNA, not charged tRNAarg, becomes the limiting factor for translation and 2) the total pool of tRNAarg is reduced (Figure 4A) so a larger fraction of the total tRNAarg can be charged by the same small amount of arginine. This experiment demonstrates how the simultaneous measurements of tRNA abundance and tRNA charging levels provides high quality information about the conditions for protein synthesis inside the cell.
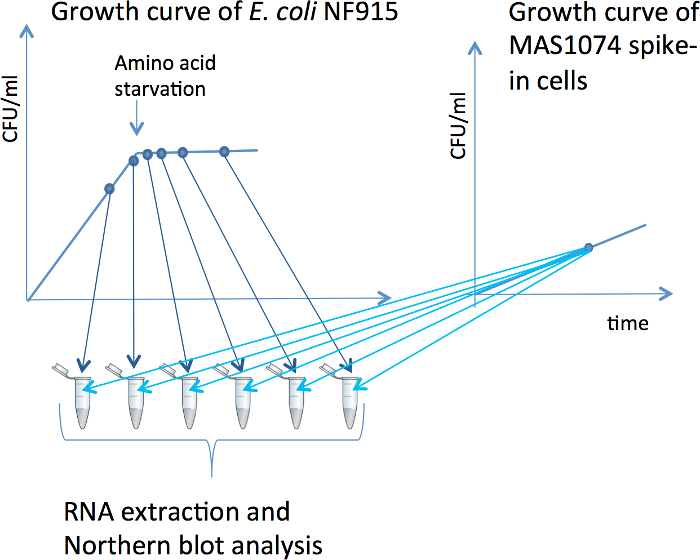
Figure 1. Addition of spike-in cells to samples. In this example, samples from a single culture of E. coli NF915 are harvested in a time series before and during amino acid starvation. A schematic of the growth curve of NF915 is shown, where the blue dots indicate the points of sample harvest. The Y-axes are log scale. Samples of the experimental culture(s) are harvested as described in Section 3. A schematic of the growth curve of the spike-in strain MAS1074 is also shown, with the point of sample harvest indicated as a blue dot. The point of the figure is to illustrate that all the experimental samples (of E. coli NF915 in this example) receive an aliquot of spike-in cells from the same reference culture. To each sample, 5% of spike-in cells are added to the experimental cells, calculated based on the OD of the cell cultures (see Section 4). Subsequently, RNA is purified from all samples and used for Northern blots. Please click here to view a larger version of this figure.
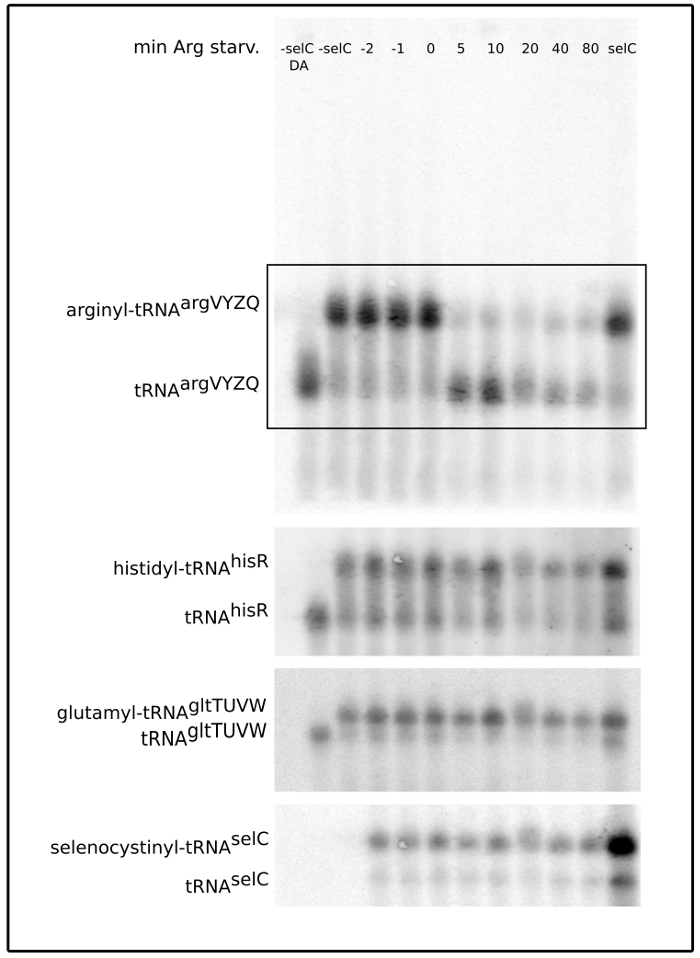
Figure 2. Phosphor imager scan of a Northern blot probed for the indicated tRNAs. The membrane contains RNA from the E. coli strain NF915 harvested at the indicated time points before and after induction of arginine starvation (minutes). Additionally, the membrane contains two lanes of samples where spike-in cells have been omitted (-selC DA and -selC), and a lane containing RNA from the spike-in cells only (selC). The -selC DA sample contains chemically deacylated tRNA. The two bands representing the charged and uncharged tRNA species are clearly separated. Note the negligible signal from tRNAselC in the lanes without added spike-in cells. The same membrane was successively stripped and reprobed for the tRNAs indicated on the left. The boxed area in the upper blot indicates the part of the blot, which is shown for the subsequent tRNA probings. Please click here to view a larger version of this figure.
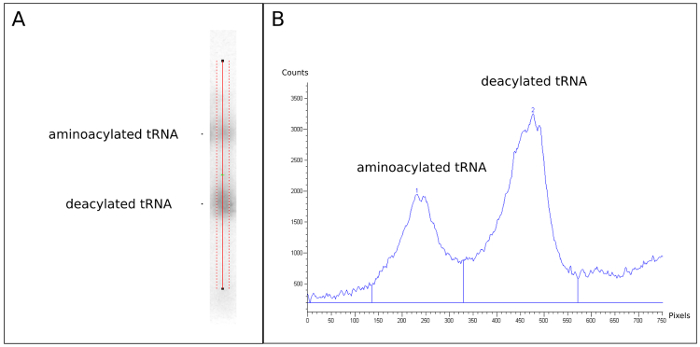
Figure 3. Quantification of Northern blot. tRNA abundance is measured using software. (A) A single lane from the Northern blot seen in Figure 2 probed for tRNAargVYZQ. The upper and lower arrow indicate the aminoacylated tRNA and the deacylated tRNA, respectively. The red lines running parallel with the lane are added and used to define the area for quantification. (B) Quantification of signal within the two broken red lines seen in (A) depicted as a graph, where the horizontal axis indicates the distance from the top of the line in (A) (in pixels), and the vertical axis shows the intensity of the signal (in counts). The two peaks containing signal originating from the aminoacylated tRNA and the deacylated tRNA, respectively, are defined manually. The data that is exported for further analysis is the value corresponding to the area under each of the two peaks. Please click here to view a larger version of this figure.
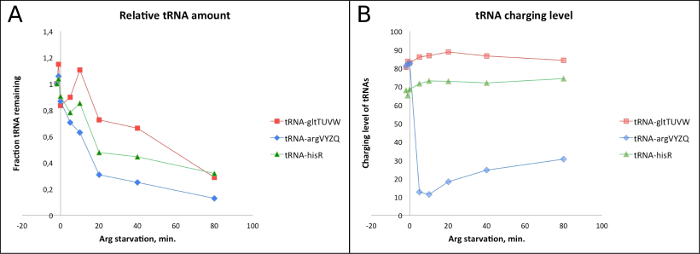
Figure 4. Representative result: Quantification of tRNA abundances and charging levels during arginine starvation. Quantification of the bands on the Northern blot shown in Figure 2. (A) The relative abundance of tRNAgltTUVW (red), tRNAargVYZQ (blue) and tRNAhisR (green). The level found at zero minutes (samples harvested just prior to the onset of starvation) is set to 1. (B) Charging level of the same three tRNAs as in (A). Please click here to view a larger version of this figure.
| Probe specificity | Probe sequence |
| tRNAargVYZQ | 5’-TCCGACCGCTCGGTTCGTAGC |
| tRNAgluTUVW | 5’-CCTGTTACCGCCGTGAAAGGG |
| tRNAhisR | 5’-CACGACAACTGGAATCACAATCC |
| tRNAleuPQVT | 5’-GTAAGGACACTAACACCTGAAGC |
| tRNAleuU | 5’-TATTGGGCACTACCACCTCAAGG |
| tRNAleuW | 5’-CTTGCGGCGCCAGAACCTAAATC |
| tRNAleuX | 5’-TATTTCTACGGTTGATTTTGAA |
| tRNAleuZ | 5’-AAAATCCCTCGGCGTTCGCGCT |
| tRNAlysQTVWYZ | 5’-TGCGACCAATTGATTAAAAGTCAAC |
| tRNAthrV | 5’-TGGGGACCTCACCCTTACCAA |
| tRNAtyrTV | 5'-TCGAACCTTCGAAGTCGATGA |
| tRNAselC | 5'-ATTTGAAGTCCAGCCGCC |
Table 1. Table of suggested probe sequences for selected tRNAs in E. coli K-12.
