Au cours des deux dernières décennies, les solides et les films fabriqués à partir de molécules organiques agrégées ont trouvé de multiples applications dans les dispositifs optoélectroniques. Les appareils basés sur ces matériaux ont de nombreuses propriétés attrayantes, y compris le faible poids, la flexibilité, la faible consommation d’énergie, et le potentiel de production bon marché en utilisant l’impression jet d’encre. Les écrans basés sur les diodes électroluminescentes organiques (OLED) remplacent les écrans cristallins liquides comme état de l’art pour les téléphones mobiles, ordinateurs portables, téléviseurs et autres appareils électroniques1,2,3,4. L’importance des OLED pour les applications d’éclairage devrait augmenter dans les années à venir4. Les performances des appareils photovoltaïques organiques s’améliorent régulièrement, avec des efficacités de conversion d’énergie supérieures à 16% récemment signalés pour les cellules solaires organiques à jonction unique5. Les matières organiques ont également le potentiel de perturber d’autres technologies, telles que les communications à fibre optique, où leur utilisation permet le développement de modulateurs électro-optiques avec des bandes passantes extrêmement élevées de 15 THz et au-dessus de6,7.
Un défi majeur dans l’optimisation des matériaux moléculaires à état solide pour les applications en optoélectronique est que généralement leurs propriétés dépendent fortement de la structure à l’échelle nanométrique du matériau. Le processus de production permet de définir la nanostructure d’un matériau dans une certaine mesure en utilisant des techniques de croissance contrôlées, telles que le dépôt de vapeur chimique,8 tentlating de molécules optiquement actives sur un autre matériau (c.-à-d., une matrice de polymère9,10), l’annealing thermique11,12, etc. Cependant, le trouble à l’échelle nanométrique est intrinsèque à la plupart des matériaux moléculaires et ne peut généralement pas être éliminé entièrement. Par conséquent, il est essentiel de comprendre comment le trouble affecte les propriétés d’un matériau et de trouver des moyens de l’ingénierie pour une performance optimale est essentiel à la conception rationnelle des matériaux optoélectroniques organiques.
Le degré de désordre dans les matériaux moléculaires est habituellement trop grand pour le traiter comme une perturbation d’une structure cristalline périodique avec une structure électronique qui peut être décrite par la théorie de bande. D’autre part, le nombre de molécules qui doivent être incluses dans une simulation pour reproduire les propriétés d’un matériau en vrac ou d’un film est trop grand pour utiliser les premiers principes des méthodes chimiques quantiques comme la théorie fonctionnelle de densité (DFT)13,14 et la théorie fonctionnelle de densité dépendante du temps (TD-DFT)15,16. Les molécules organiques avec des applications en optoélectronique ont généralement des systèmes relativement grands – conjugués; beaucoup ont également des groupes de donateurs et d’accepteur. Capturer le comportement correct de transfert de charge dans de telles molécules est essentiel pour calculer leurs propriétés optoélectroniques, mais il ne peut être accompli en utilisant des fonctions hybrides corrigées à longue portée dans TD-DFT17,18,19,20. Les calculs qui utilisent ces fonctions s’érigent super linéairement avec la taille du système et, à l’heure actuelle, ils ne sont pratiques que pour modéliser les propriétés optoélectroniques de molécules organiques individuelles ou de petits agrégats moléculaires qui peuvent être décrits en utilisant pas plus de 104 fonctions de base atomique. Une méthode de simulation qui pourrait décrire les matériaux désordonnés qui se composent d’un grand nombre de chromophores serait très utile pour la modélisation de ces systèmes.
L’ampleur des interactions intermoléculaires dans les matériaux moléculaires est souvent comparable ou plus petite que l’ordre de variation des paramètres énergétiques (comme les énergies eigenstate ou les énergies d’excitation) entre les molécules individuelles qui composent le matériau. Dans de tels cas, la modélisation multi-échelle est l’approche la plus prometteuse pour comprendre et optimiser les propriétés optoélectroniques des grands systèmes moléculaires désordonnés21,22,23. Cette approche utilise des méthodes chimiques quantiques de premier plan (généralement DFT et TD-DFT) pour calculer avec précision les propriétés des molécules individuelles qui composent le matériau. Le Hamiltonien d’un échantillon matériel suffisamment grand pour représenter le matériau moléculaire en vrac (peut-être, en utilisant des conditions limites périodiques) est ensuite construit à l’aide des paramètres qui ont été calculés pour les molécules individuelles. Ce Hamiltonien peut ensuite être utilisé pour calculer les paramètres optoélectroniques d’un grand agrégat moléculaire, d’un film mince ou d’un matériau moléculaire en vrac.
Les modèles Exciton sont une sous-classe de modèles multi-échelles dans lesquels les états excités d’un matériau moléculaire sont représentés dans une base d’excitons: paires de trous d’électrons qui sont liés par l’attraction Coulomb24,25. Pour la modélisation de nombreux processus d’état excités, il suffit d’inclure uniquement les excitons Frenkel26, où l’électron et le trou sont localisés sur la même molécule. Les excitons de transfert de charge, où l’électron et le trou sont localisés sur différentes molécules, peuvent devoir être inclus dans certains cas (p. ex., lors de la séparation des charges de modélisation dans les systèmes de donneur-accepteur)27,,28. Bien que les modèles d’exciton soient des modèles à plusieurs échelles qui peuvent être paramétrisés en utilisant uniquement des calculs de principe sur des molécules individuelles, ils tiennent toujours compte des interactions intermoléculaires. Les deux principaux types d’interaction qu’ils peuvent expliquer sont (a) des accouplements excitoniques entre les molécules qui caractérisent la capacité des excitons à délocaliser ou à transférer entre les molécules et (b) la polarisation électrostatique de la densité d’électrons sur une molécule par la distribution de charge sur les molécules environnantes. Nous avons déjà montré que ces deux facteurs sont importants pour la modélisation des propriétés optiques et électro-optiques des agrégats moléculaires, tels que le spectre d’absorption optique29 et les premières hyperpolarizabilities30.
Dans cet article, nous présentons un protocole pour la parmétrisation des modèles d’exciton qui peuvent être utilisés pour calculer les spectres optiques et d’autres propriétés optoélectroniques de grands agrégats moléculaires et de matériaux moléculaires en vrac. L’excitonique Hamiltonian est supposé être un Hamiltonian serré-contraignant24,25,
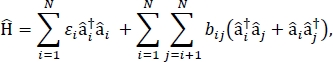
où je suis l’énergiei d’excitation de la molécule ith dans le matériau, bij est le couplage excitonique entre le ie et les molécules je, â i– et â je suis la création et les opérateurs d’anéantissement, respectivement, pour un état excité sur la molécule ie dans le matériau. Les paramètres accitoniques de Hamiltoniens sont trouvés à l’aide de calculs TD-DFT qui sont effectués sur des molécules individuelles qui composent le matériau. Dans ces calculs TD-DFT, la distribution de charge sur toutes les autres molécules du matériau est représentée par l’intégration électrostatique des charges de points atomiques pour tenir compte de la polarisation électrostatique de la densité électronique d’une molécule. Les énergies d’excitation,i,pour les molécules individuelles sont prises directement à partir de la sortie de calcul TD-DFT. Les accouplements excitoniques, bij, entre les molécules sont calculés à l’aide de la méthode cube de densité de transition31, avec les densités de transition d’état au sol pour excitées pour les molécules en interaction prélevées à partir de la sortie d’un calcul TD-DFT dans Gaussian32 et post-traitement à l’aide de l’analyseur multifonctionnel multifonctionnel multivolction multifunction33. Pour simuler les propriétés des solides moléculaires en vrac, des conditions limites périodiques peuvent être appliquées au Hamiltonien.
Le protocole actuel exige que l’utilisateur ait accès aux programmes Gaussian32 et Multiwfn33. Le protocole a été testé à l’aide de Gaussian 16, révision B1 et Multiwfn version 3.3.8, mais devrait également fonctionner pour d’autres versions récentes de ces programmes. En outre, le protocole utilise un utilitaire personnalisé C et un certain nombre de scripts personnalisés python 2.7 et Bash, dont le code source est fourni sous la licence publique générale GNU (Version 3) à https://github.com/kocherzhenko/ExcitonicHamiltonian. Les calculs sont destinés à être effectués sur une machine exécutant un système d’exploitation de la famille Unix/Linux.
