За последние два десятилетия твердые вещества и пленки, изготовленные из агрегированных органических молекул, нашли несколько применений в оптоэлектронных устройствах. Устройства, основанные на таких материалах, обладают многими привлекательными свойствами, включая небольшой вес, гибкость, низкое энергопотребление и потенциал для дешевого производства с использованием струйной печати. Дисплей на основе органических светоизлучающих диодов (OLEDs) заменяют жидкие кристаллические дисплеи, как современные для мобильных телефонов, ноутбуков, телевизоров и других электронных устройств1,2,3,4. Важность OLED для освещения приложений, как ожидается, возрастет в ближайшие годы4. Производительность органических фотоэлектрических устройств неуклонно улучшается, с эффективностью преобразования энергии выше 16% недавно сообщили для одноразового соединения органических солнечных элементов5. Органические материалы также имеют потенциал, чтобы нарушить другие технологии, такие как волоконно-оптические коммуникации, где их использование позволяет развитие электрооптических модуляторов с чрезвычайно высокой пропускной способностью 15 ТГц и выше6,7.
Основная проблема в оптимизации твердотельных молекулярных материалов для применения в оптоэлектронике заключается в том, что, как правило, их свойства сильно зависят от наномасштабной структуры материала. Производственный процесс позволяет определить наноструктуру материала в некоторой степени с помощью контролируемых методов роста, таких как химическое осаждение пара,8 шаблонов оптически активных молекул на другой материал (т.е. полимерную матрицу9,10), тепловое аннулирование11,12и т.д. Тем не менее, наноразмерное расстройство присуще большинству молекулярных материалов и, как правило, не может быть полностью устранено. Таким образом, понимание того, как расстройство влияет на свойства материала и найти способы инженера его для оптимальной производительности имеет важное значение для рационального проектирования органических оптоэлектронных материалов.
Степень расстройства в молекулярных материалах, как правило, слишком велика, чтобы рассматривать его как возмущение периодической кристаллической структуры с электронной структурой, которая может быть описана теорией группы. С другой стороны, количество молекул, которые должны быть включены в моделирование, чтобы воспроизвести свойства объемного материала или пленки слишком велика, чтобы использовать первые принципы квантовых химических методов, таких как плотность функциональной теории (DFT)13,14 и зависит от времени плотность функциональной теории (TD-DFT)15,16. Органические молекулы с применением в оптоэлектронике, как правило, имеют относительно большие конъюгированные системы; многие из них также имеют группы доноров и принимающих. Захват правильного поведения передачи заряда в таких молекулах имеет важное значение для расчета их оптоэлектронных свойств, но это может быть достигнуто только с помощью дальнего скорректированных гибридных функций в TD-DFT17,18,19,20. Расчеты, которые используют такие функциональные масштабы супер линейно с размером системы и, в настоящее время, они только практичны для моделирования оптоэлектронных свойств отдельных органических молекул или небольших молекулярных агрегатов, которые могут быть описаны с использованием не более чем 104 атомных базовых функций. Метод моделирования, который мог бы описать неупорядоченные материалы, которые состоят из большого количества хромофор было бы очень полезно для моделирования этих систем.
Величина межмолекулярных взаимодействий в молекулярных материалах часто сравнима с порядком или меньше, чем порядок изменения энергетических параметров (таких как энергии эйгенстата или энергии возбуждения) между отдельными молекулами, которые составляют материал. В таких случаях многомасштабное моделирование является наиболее перспективным подходом к пониманию и оптимизации оптоэлектронных свойств крупных неупорядоченных молекулярных систем21,,22,,23. Этот подход использует первопринципные квантовые химические методы (обычно DFT и TD-DFT) для точного расчета свойств отдельных молекул, которые составляют материал. Гамильтониан из материала образца, который является достаточно большим, чтобы представлять объеммолекулярного материала (возможно, с использованием периодических условий границы) затем построен с использованием параметров, которые были рассчитаны для отдельных молекул. Этот Гамильтониан может быть использован для расчета оптоэлектронных параметров большого молекулярного агрегата, тонкой пленки или оптово-молекулярного материала.
Exciton модели подкласса многомасштабных моделей, в которых возбужденные состояния молекулярного материала представлены в основе эксцитонов: электронно-отверстие пары, которые связаны Coulomb притяжения24,25. Для моделирования многих возбужденных государственных процессов достаточно включить только френкель эксцтоны26, где электрон и отверстие локализуются на той же молекуле. В некоторых случаях (например, при моделировании разделения заряда в системах донора-приемного органа, когда электрон и отверстие локализованы на различных молекулах, могут потребоваться включить их (например, при моделировании разделения заряда в системах донора-приемного лица)27,28. Хотя модели exciton являются многоразмерными моделями, которые могут быть параметризированы, используя только расчеты по принципу первого принципа на отдельных молекулах, они по-прежнему учитывают межмолекулярные взаимодействия. Два основных типа взаимодействия, которые они могут объяснить, являются а) возбуждающие соединения между молекулами, которые характеризуют способность экзитонов к делокализации или передачи между молекулами и (b) электростатической поляризации плотности электрона на молекуле распределением заряда на окружающих молекулах. Ранее мы показали, что оба этих фактора важны для моделирования оптических и электрооптических свойств молекулярных агрегатов, таких как оптическая спектра поглощения29 и первая гиперполярная30.
В этой работе мы представляем протокол для параметрионирующих моделей exciton, которые могут быть использованы для расчета оптических спектров и других оптоэлектронных свойств крупных молекулярных агрегатов и объемных молекулярных материалов. Возбуждающий Гамильтониан считается плотно обязательным Hamiltonian24,25,
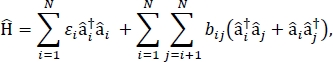
где я энергию возбуждения ith в материале, bij excitonic соединение между ith и молекулами jth, iq и я операторы творения и аннигиляции, соответственно, для возбужденного положения на ith молекуле в материале. Возбужданые параметры Гамильтона обнаруживаются с помощью расчетов TD-DFT, которые выполняются на отдельных молекулах, которые составляют материал. В этих расчетах TD-DFT распределение заряда на всех других молекулах материала представлено электростатическим встраиванием атомных точечных зарядов для учета электростатической поляризации электронной плотности молекулы. Энергии возбуждения, я,iдля отдельных молекул взяты непосредственно из tD-DFT выход расчета. Возбужданые соединения, bij, между молекулами рассчитываются с помощьюметодакубической плотности перехода перехода перехода от земли к возбуждению для взаимодействующих молекул, взятых из вывода расчета TD-DFT в Gaussian32 и после обработки с помощью многофункционального анализа тауэра Multiwfn 33. Для имитации свойств навалочных молекулярных твердых веществ, периодические пограничные условия могут быть применены к Гамильтониан.
Текущий протокол требует, чтобы пользователь имеет доступ к Гауссиан32 и Multiwfn33 программ. Протокол был протестирован с использованием Gaussian 16, пересмотра Версии B1 и Multiwfn 3.3.8, но также должен работать для других последних версий этих программ. Кроме того, в протоколе используется пользовательская утилита C и ряд пользовательских скриптов python 2.7 и Bash, исходный код которого предоставляется в соответствии с GNU General Public License (версия 3) в https://github.com/kocherzhenko/ExcitonicHamiltonian. Расчеты предназначены для выполнения на машине, работающей на операционной системе семейства Unix/Linux.
