Analysis of TGF-β signaling
The key step in TGF-β signaling is the phosphorylation of the two most carboxy terminal serine residues in the SSXS motif (Figure 2) by TβRI kinase38,39. To investigate TGF-β signaling responses, we performed Western blotting of phosphorylated SMAD2. In the premalignant human breast MCF10A-Ras cells, the phosphorylation of SMAD2 significantly increased in response to TGF-β stimulation for 1 hour, while the expression of total SMAD2/3 was not affected by TGF-β treatment (Figure 4A). By using the TGF-β-induced SMAD3/4-driven CAGA-luc transcriptional reporter assay, we found that TGF-β markedly induced the luciferase reporter in the MCF10A-Ras cells line compared to non-treated cells (Figure 4B). Moreover, we observed that well-characterized direct transcriptional gene targets of TGF-β including SMAD7 and SERPINE1 (encoding the PAI-1 protein), were highly expressed in TGF-β-treated MCF10A-Ras cells (Figure 4C).
Analysis of TGF-β-induced EMT
We assessed TGF-β-induced EMT with various methods, such as morphological changes, the expression of EMT markers at the mRNA and protein levels and immunofluorescence staining of EMT markers36. NMuMG epithelial cells treated with TGF-β for 1 and 2 days changed from a classic epithelial morphology to a spindle-shaped mesenchymal-like morphology, as shown by phase contrast microscopy (Figure 5A). Consistent with the morphological changes, we observed that TGF-β treatment led to an increase in the protein expression of mesenchymal markers, including N-cadherin, Snail, and Slug37 (Figure 5B). In contrast, E-cadherin, an epithelial marker, was downregulated in NMuMG cells after 2 days of TGF-β treatment (Figure 5B). In addition, we performed quantitative real-time-polymerase chain reaction (qRT-PCR) to investigate the gene expression of EMT markers. CDH1 (encoding the E-cadherin protein) was significantly decreased, while mesenchymal markers such as SNAIL and Zinc finger E-box-binding homeobox 2 (ZEB2) were markedly increased after TGF-β stimulation in NMuMG cells compared to untreated cells (Figure 5C). TGF-β-induced EMT in NMuMG cells was further confirmed by immunofluorescence staining of E-cadherin. Upon TGF-β stimulation for 2 days, NMuMG cells expressed less E-cadherin than cells in the uninduced control group, as analyzed by confocal microscopy (Figure 5D). Moreover, NMuMG cells in the presence of TGF-β formed more actin stress fibers, as shown by confocal microscopy (Figure 5E).
SB431542 and GW788388 inhibit TGF-β signaling and TGF-β-induced EMT
SB431542 is an ATP competitive inhibitor of the kinase domain of TβRI, also termed activin receptor-like kinase 5 (ALK5), while GW788388 inhibits TβRI and TβRII kinase activity. Both inhibitors can inhibit TGF-β receptor signaling40. Thus, we treated NMuMG cells with different concentrations of GW788388 in the presence of TGF-β for 1 hour. As expected, GW788388 inhibited TGF-β-induced SMAD2 phosphorylation in a dose-dependent manner (Figure 6A). Additionally, the TGF-β-mediated phosphorylation of SMAD2 was blocked by SB431542 treatment (Figure 6A). Phosphorylated SMAD2/3 forms a heteromeric complex with SMAD4 and translocates into the nucleus to modulate the transcription of target genes. Therefore, we investigated the translocation of SMAD2/3 in NMuMG cells by immunofluorescence staining of SMAD2/3. The data demonstrated that both SB431542 and GW788388 significantly inhibited the TGF-β-induced nuclear translocation and accumulation of SMAD2/3 in NMuMG cells (Figure 6B). Furthermore, the inhibitory effects of SB431542 and GW788388 were also observed in the mRNA expression levels of important TGF-β target genes involved in EMT, including PAI-1, SNAIL, E-cadherin and Fibronectin (Figure 6C). These data suggested that SB431542 and GW788388 blocked TGF-β signaling and TGF-β-induced EMT.
Analysis of the transdifferentiation of mesenchymal breast cancer cells into adipocytes
EMT plays a vital role in enhancing cellular plasticity in cancers and results in the development of therapy resistance. Cancer cell plasticity can be directly targeted and inhibited with a trans-differentiation approach, such as forced adipogenesis30. We used Py2T murine breast cancer cells, which were derived from the mammary gland of a mouse mammary tumor virus-polyoma middle tumor-antigen (MMTV-PyMT) transgenic mouse, as a cellular model of EMT-induced cancer cell plasticity. Based on an established protocol41, we treated EMT-derived Py2T murine breast cancer cells with the anti-diabetic drug rosiglitazone for 10 days to induce adipogenesis. Adipogenesis was assessed by visualizing lipid droplets using oil red O staining. Fat cells were readily detected in rosiglitazone-treated Py2T murine breast cancer cells (Figure 7), which demonstrated that treatment with rosiglitazone alone is sufficient to promote the transdifferentiation of EMT-derived breast cancer cells into adipocytes.
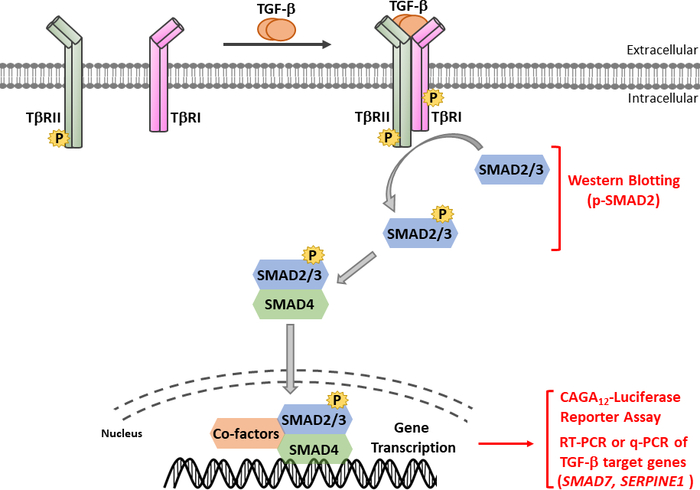
Figure 1: TGF-β/SMAD signaling. TGF-β signaling initiates with the binding of TGF-β to TGF-β type II receptor (TβRII), a constitutively active kinase, that phosphorylates TGF-β type I receptor (TβRI). Then, activated TβRI kinase phosphorylates SMAD2/3. A peptide containing the SSXS motif of SMAD2 with two carboxy terminal phosphorylated serine residues was used to obtain polyclonal antisera recognizing phosphor-SMAD2 (p-SMAD2). Therefore, the analysis of p-SMAD2 expression by Western blotting can be used to determine the activation of the TGF-β signaling pathway. Phosphorylated SMAD2/3 can form heteromeric complexes with SMAD4, which then translocate into the nucleus to modulate transcriptional responses. The CAGA12-luciferase reporter assay and quantitative real time PCR (qRT-PCR) for the mRNA expression of TGF-β target genes such as SMAD7 and SERPINE1 (encoding PAI-1 protein), can be used to analyze TGF-β-induced SMAD3-dependent transcriptional responses. Please click here to view a larger version of this figure.
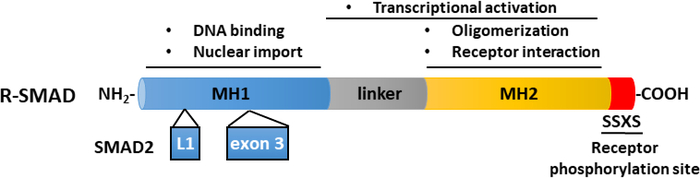
Figure 2: Schematic structure of R-SMADs (SMAD2 and SMAD3). The MH1 (blue) and MH2 (yellow) domains are conserved among R-SMADs, but the linker region (gray) is not conserved. The MH1 domain of SMAD3 harbors a DNA-binding motif, while SMAD2 cannot directly bind DNA, because of an insertion (exon 3) in its MH1 domain. The MH2 domain mediates SMAD oligomerization, interaction with TGF-β receptors, and protein binding and is involved in transcriptional regulation. SMAD2 and SMAD3 can be activated by the phosphorylation of the SSXS motif (in red) in their C termini. Please click here to view a larger version of this figure.
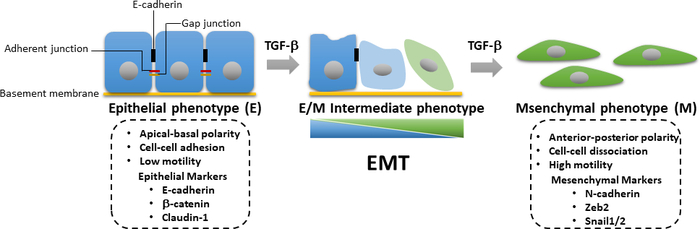
Figure 3: TGF-β-induced EMT. During TGF-β-induced epithelial–mesenchymal transition (EMT), the cells undergo loss of epithelial and acquisition of mesenchymal characteristics with enhanced cell motility and invasion ability. The induction of EMT leads to the expression of mesenchymal markers such as N-cadherin, Zeb2, and Snail1/2, as well as the downregulation of epithelial markers including E-cadherin, β-catenin, and claudin-1. The accumulated loss or gain of epithelial/mesenchymal (E/M) characteristics causes a cell to enter intermediate states in a reversible manner. Please click here to view a larger version of this figure.
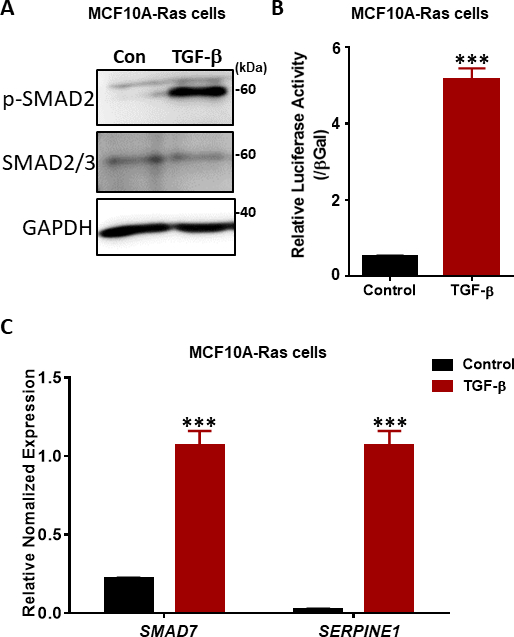
Figure 4: TGF-β signaling responses in MCF10A-Ras cells. (A) MCF10A-Ras cells were treated either with or without TGF-β (2.5 ng/mL) for 1 hour, and cell lysates were immunoblotted for phosphorylated SMAD2 (p-SMAD2), total SMAD2/3 and GAPDH (as a loading control). The size marker is shown on the right. Con: Control group without TGF-β treatment. (B) Analysis of TGF-β (5 ng/mL) activity using the SMAD3–SMAD4-dependent CAGA12-luciferase (LUC) transcriptional reporter in MCF10A-Ras cells. The values are normalized to β-galactosidase (βGal) activity. Data are expressed as the mean ± s.d, n = 3. Student’s t test, ***P ≤ 0.001 (C) qRT-PCR analysis of the TGF-β target genes SMAD7 and SERPINE1 (encoding the PAI-1 protein) in MCF10A-Ras cells treated with TGF-β (2.5 ng/mL) for 6 hours. GAPDH was used as an internal control. Data are expressed as the mean ± s.d, n = 3. Student’s t test, ***P ≤ 0.001. Please click here to view a larger version of this figure.
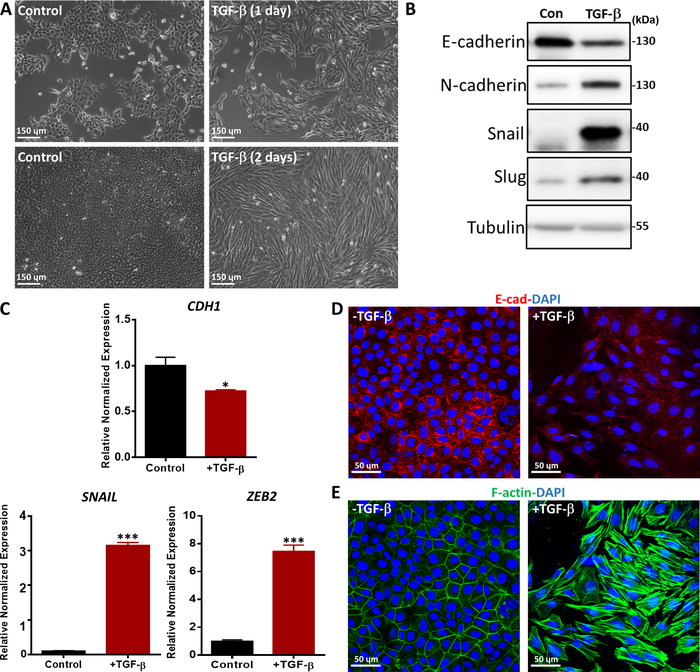
Figure 5: TGF-β-induced EMT in NMuMG cells. (A) Morphology of NMuMG cells treated with TGF-β (2.5 ng/mL) for 1 or 2 days. In the presence of TGF-β, NMuMG cells transdifferentiated into a mesenchymal phenotype. Scale bar = 150 µm (B) NMuMG cells were treated with or without TGF-β (5 ng/mL) for 2 days, and EMT markers were analyzed by Western blotting. The size marker is as indicated on the right. Con: Control group without TGF-β treatment. (C) Gene expression analysis of EMT markers (CDH1 (encoding the E-cadherin protein), SNAIL and ZEB2) in NMuMG cells treated for 2 days with TGF-β (5 ng/mL). GAPDH was used as an internal control. The results are expressed as the mean ± s.d., n = 3. Student’s t-test, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. (D) NMuMG cells were stained by immunofluorescence to detect the expression of the epithelial marker E-cadherin (red) after TGF-β (2.5 ng/mL) treatment for 2 days. Nuclei were counterstained with DAPI (blue). Images were captured with confocal microscopy. Scale bar = 50 µm (E) NMuMG cells were stained with fluorescein-phalloidin (green) to visualize F-actin. Nuclei were counterstained with DAPI (blue). Scale bar = 50 µm. Please click here to view a larger version of this figure.
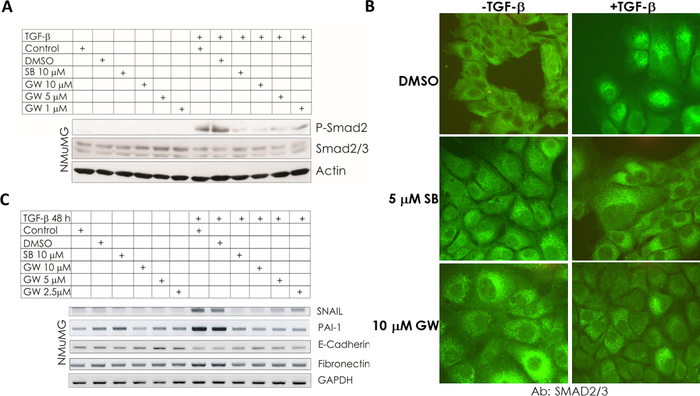
Figure 6: TGF-βsignaling and TGF-β-induced EMT were inhibited by SB431542 and GW788388. (A) NMuMG cells were treated with 10 µM of SB431542 (SB) or the indicated concentrations of GW788388 (GW) in the presence or absence of TGF-β (5 ng/mL) for 1 hour. The cell lysates were immunoblotted for p-SMAD2, SMAD2/3 and GAPDH. (B) NMuMG cells were treated with 5 µM of SB431542 (SB) or 10 µM of GW788388 (GW) in the presence or absence of 5 ng/mL of TGF-β for 1 hour and stained by immunofluorescence to detect the nuclear translocation of SMAD2/3 (green). Images were captured with confocal microscopy. (C) Expression of TGF-β target genes, including PAI-1 and genes encoding EMT markers, including SNAIL, E-Cadherin and Fibronectin, were analyzed by reverse transcriptase polymerase chain reaction (RT-PCR) in NMuMG cells after SB or GW treatment and TGF-β stimulation for 48 hours. GAPDH served as a loading control. Control denotes nontreated cells. This figure has been modified from Petersen M. et al. 34 with permission from publisher. Please click here to view a larger version of this figure.
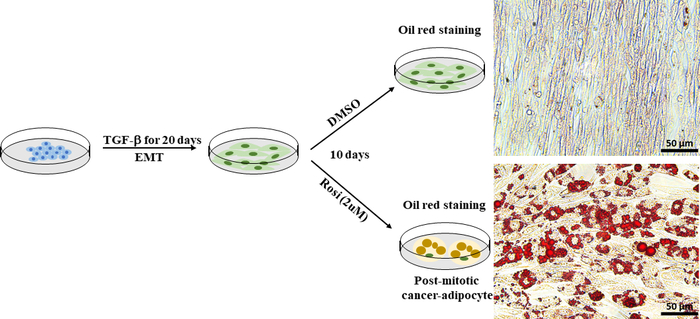
Figure 7: EMT-derived Py2T murine breast cancer cells can be induced to differentiate into adipocytes. Py2T murine breast cancer cells were stimulated with 2 ng/mL TGF-β for 20 days to induce complete EMT. Then, the cells were treated either with DMSO as a vehicle control or rosiglitazone (2 µM) for 10 days to allow the differentiation of mesenchymal cancer cells and induce adipogenesis. The medium was changed every 2 days. After 10 days of treatment, cells were stained with oil red O. Scale bar = 50 µm. Please click here to view a larger version of this figure.
| Species | Gene name | Forward (5' to 3') | Reverse (5' to 3') | ||
| Human | GAPDH | TGCACCACCAACTGCTTAGC | GGCATGGACTGTGGTCATGAG | ||
| SMAD7 | TCCAGATGCTGTGCCTTCC | GTCCGAATTGAGCTGTCCG | |||
| SERPINE1 | CACAAATCAGACGGCAGCACT | CATCGGGCGTGGTGAACTC | |||
| Mouse | GAPDH | TGGCAAAGTGGAGATTGTTGCC | AAGATGGTGATGGGCTTCCCG | ||
| CDH1 | ACCAAAGTGACGCTGAAGTC | GAGGATGTACTTGGCAATGG | |||
| SNAIL | CAGCTGGCCAGGCTCTCGGT | GCGAGGGCCTCCGGAGCA | |||
| ZEB2 | TTCTGCAAGCCTCTGTAGCC | TTCTGGCCCCATTGCATCAT | |||
Table 1: Primers used for qRT-PCR.
