Labelling and Visualization of Mitochondrial Genome Expression Products in Baker’s Yeast Saccharomyces cerevisiae
Summary
Baker’s yeast mitochondrial genome encodes eight polypeptides. The goal of the current protocol is to label all of them and subsequently visualize them as separate bands.
Abstract
Mitochondria are essential organelles of eukaryotic cells capable of aerobic respiration. They contain circular genome and gene expression apparatus. A mitochondrial genome of baker’s yeast encodes eight proteins: three subunits of the cytochrome c oxidase (Cox1p, Cox2p, and Cox3p), three subunits of the ATP synthase (Atp6p, Atp8p, and Atp9p), a subunit of the ubiquinol-cytochrome c oxidoreductase enzyme, cytochrome b (Cytb), and mitochondrial ribosomal protein Var1p. The purpose of the method described here is to specifically label these proteins with 35S methionine, separate them by electrophoresis and visualize the signals as discrete bands on the screen. The procedure involves several steps. First, yeast cells are cultured in a galactose-containing medium until they reach the late logarithmic growth stage. Next, cycloheximide treatment blocks cytoplasmic translation and allows 35S methionine incorporation only in mitochondrial translation products. Then, all proteins are extracted from yeast cells and separated by polyacrylamide gel electrophoresis. Finally, the gel is dried and incubated with the storage phosphor screen. The screen is scanned on a phosphorimager revealing the bands. The method can be applied to compare the biosynthesis rate of a single polypeptide in the mitochondria of a mutant yeast strain versus the wild type, which is useful for studying mitochondrial gene expression defects. This protocol gives valuable information about the translation rate of all yeast mitochondrial mRNAs. However, it requires several controls and additional experiments to make proper conclusions.
Introduction
Mitochondria are the organelles deeply involved in the metabolism of a eukaryotic cell. Their electron transfer chain supplies the cell with ATP, the main energetic currency used in multiple biochemical pathways. Besides, they are involved in apoptosis, fatty acid and heme synthesis, and other processes. Dysfunction of mitochondria is a well-known source of human disease1. It can result from mutations in nuclear or mitochondrial genes encoding structural or regulatory components of the organelles2. Baker’s yeast Saccharomyces cerevisiae is an excellent model organism for studying mitochondrial gene expression due to several reasons. First, their genome is completely sequenced3, well-annotated, and a big sum of data is already available in literature thanks to the long history of investigations carried out with this organism. Second, the manipulations with their nuclear genome are relatively fast and easy because of their fast growth rate and highly efficient homologous recombination system. Third, baker’s yeast S. cerevisiae is one of the few organisms for which the manipulations with mitochondrial genomes are developed. Finally, baker's yeast is an aerobe-anaerobe facultative organism, which allows isolation and study of respiratory defective mutants, since they can grow in media containing fermentable carbon sources.
We describe the method to study mitochondrial gene expression of baker’s yeast S. cerevisiae at the translational level4. Its main principle comes from several observations. First, the yeast mitochondrial genome encodes only eight proteins: three subunits of the cytochrome c oxidase (Cox1p, Cox2p, and Cox3p), three subunits of the ATP synthase (Atp6p, Atp8p, and Atp9p), a subunit of the ubiquinol-cytochrome c oxidoreductase enzyme, cytochrome b (Cytb), and mitochondrial ribosomal protein Var1p5. This number is small, and all of them can be separated by electrophoresis on a single gel in the appropriate conditions. Second, mitochondrial ribosomes belong to the prokaryotic class rather than eukaryotic6, and therefore, the sensitivity to antibiotics is different for yeast cytoplasmic and mitochondrial ribosomes. It allows the inhibition of cytoplasmic translation with cycloheximide, providing the conditions when the labeled amino acid (35S-methionine) is incorporated only in mitochondrial translation products. As a result, the experiment gives information about the rate of amino acid incorporation in mitochondrial proteins synthesized de novo, reflecting the overall efficiency of mitochondrial translation for each of the eight products
Protocol
1. Yeast culture preparation
- Streak yeast from the frozen stock cultures on fresh plates with the appropriate medium. Put the plates in a culture incubator at 30 °C for 24–48 h.
NOTE: Let the temperature-sensitive mutants grow at the permissive temperature. - Inoculate yeast cultures in 2 mL of YPGal medium (2% peptone, 1% yeast extract, 2% galactose) from the fresh streak in 15 mL tubes and incubate them overnight agitating at 200 rpm at 30 °C.
- Measure the optical density of the culture at a wavelength of 600 nm (OD600).
- Take the volume corresponding to 0.2 absorbance units in sterile tubes, pellet yeast cells at 9,000 x g for 30 s at room temperature, and discard the supernatant.
- Wash cells with 0.5 mL of sterile water by vortexing for 5 s. Pellet yeast cells at 9,000 x g for 30 s at room temperature and discard the supernatant. Dilute cells in 2 mL of fresh YPGal medium.
- Incubate agitating at 200 rpm and 30 °C until OD600 reaches 1.5–1.9 values.
NOTE: Yeast growth rates vary, so be prepared to wait. It is reasonable to make steps 1.3–1.6 early in the morning. It usually takes 4–5 h, but can take longer.
2. Radioactive isotope incorporation
- Transfer the culture volume equivalent to one optical unit in a microcentrifuge tube. Spin the tubes at 3,000 x g for 1 min and discard the supernatant. Wash with 0.5 mL of sterile water by vortexing for 5 s.
- Pellet yeast cells at 9,000 x g for 30 s at room temperature and discard the supernatant. Resuspend yeast cells in 0.5 mL of sterile translation buffer. Place the suspension in a 15 mL tube.
NOTE: Translation buffer is a solution containing 2% galactose (w/v) and 50 mM potassium phosphate with pH 6.0.
- Pellet yeast cells at 9,000 x g for 30 s at room temperature and discard the supernatant. Resuspend yeast cells in 0.5 mL of sterile translation buffer. Place the suspension in a 15 mL tube.
- Add cycloheximide to cell suspension up to a final concentration of 0.2 mg/mL. Incubate for 5 min agitating at 200 rpm and 30 °C to inhibit cytosolic translation.
NOTE: Cycloheximide solution (20 mg/mL, in ethanol) should be prepared fresh before the experiment. Chloramphenicol can be successfully substituted with anisomycin in the same concentration7. - Add 25–30 μCi of 35S-methionine to the cell suspension and incubate for 30 min agitating at 200 rpm and 30 °C.
NOTE: The mixture of 35S-methionine and 35S-cysteine can also be used. Pure 35S-methionine results in the strongest signal and the best signal-to-noise ratio. Generally, methionine is more effective than cysteine because the content of this amino acid in mitochondrial proteins is higher. However, the EasyTag mixture of 35S-methionine and cysteine is less expensive and gives comparable results7.
CAUTION: 35S-methionine is radioactive. Follow the usual safety practices for handling radioactive materials.
NOTE: It is well known that the incorporation of radioactivity reaches a limit due to which the total signals level off over time. Once this limit is reached, it is almost impossible to determine the rates by which the different translation products are synthesized. To analyze the rate of mitochondrial translation, it is imperative to be in a condition in which the signal intensity increases with time of incubation with 35S-methionine in a linear manner. For common laboratory wild-type strains, the signal is already saturated for incubation times far shorter than the 30 min. Translational mutants can behave very differently. A time-course should be performed to establish the kinetics of radioactivity incorporation and thus the rate of translation, at least when working with a new strain. For this, we suggest taking samples with an interval of several minutes (e.g., 2.5, 5.0, 7,5, 10, and 20 min). - Add unlabeled "cold" methionine (final concentration should be 20 mM) and puromycine (final concentration should be 1–10 μg/mL) to stop the labeling. Incubate for 10 min agitating at 200 rpm and 30 °C.
NOTE: This step is critical to give ribosomes time to finish translating the peptides. Otherwise, all polypeptides shorter than full-length will be detected at a different size. A time-course experiment (pulse-chase) can be done at this step to determine the stability of mitochondrial translation products. For this, continue the incubation taking samples with an interval of 30 min (e.g., 30, 60, 90, and 120 min).
3. Yeast cell lysis and extraction of proteins
- Collect yeast cells by centrifugation at 9,000 x g for 30 s. Wash the cells with 0.5 mL of sterile water by vortexing for 5 s. Pellet yeast cells at 9,000 x g for 30 s at room temperature. Discard the supernatant.
NOTE. The protocol can be paused here. Store the samples at -20 °C. - Add 75 μL of lysis buffer to the pellet and vortex for 5–10 s.
NOTE: Lysis buffer is a solution of 1.8 M NaOH, 1 M β-mercaptoethanol, and 1 mM PMSF in water. Avoid excessive incubation with lysis buffer leading to alkaline hydrolysis of proteins. Immediately proceed to step 3.3. - Add 500 μL of 0.5 M Tris-HCl buffer with pH 6.8. Vortex briefly.
4. Precipitation of proteins
CAUTION: Methanol and chloroform are organic solvents. Follow the usual safety practices for handling organic substances.
- Add 600 µL of methanol to the sample. Vortex for 5 s.
- Add 150 µL of chloroform to the sample. Vortex for 5 s.
- Centrifuge the samples for 2 min at 12,000 x g. Carefully discard the upper phase with a sampler.
- Add 600 µL of methanol to the sample. Mix carefully by inverting the tube several times.
- Centrifuge the samples for 2 min at 12,000 x g. Discard the supernatant.
- Air-dry the pellet for 2 min at 80 °C.
NOTE: All of the liquid should evaporate. There is a risk to have an aberrant separation of proteins in the gel if the pellet was dried insufficiently. However, it is not possible to over-dry the pellet, so it can even be stored overnight at 4 °C. - Dissolve precipitated proteins in 60 µL of 1x Laemmli sample buffer.
- Heat for 10 min at 40 °C.
NOTE: Avoid boiling the samples at 95 °C, because this causes aggregation. If the aggregates still form, Spin the samples at 12,000 x g for 2 min and collect the supernatant. Samples can be stored at -20 °C. The protocol can be paused here.
5. SDS-PAGE
- Cast the 17.5% Laemmli SDS-polyacrylamide gel.
NOTE: 6 M urea can be added to the gel to resolve better atp8 and atp9. Gradient 15%–20% gels can also give better resolution. - Load 15 μL (40–50 µg) of each sample in the pockets.
NOTE: It is not necessary to measure protein concentration if OD600 values were close in step 1.6. If not, measure protein concentrations using the assay with detergent-compatible reagents. - Run the gel in a cold room until the blue dye reaches approximately 65% of the gel length.
NOTE: For the system used (e.g., Protean II xi cell), we use either of the two modes: 16–17 h at 5 V/cm or 5 h at 15 V/cm. No proteins run faster than the bromophenol blue dye, but long runs can result in blurring of atp8 and atp9 signals. - Stain the gel with Coomassie brilliant blue and make a scan or photo, which is required as a loading control.
NOTE: An alternative way to make a loading control is immunoblotting with an antibody to mitochondrial “house-keeping” gene, e. g., porin 1.
6. Autoradiography
- Dry the gel in a gel-dryer. Keep it in the cassette with a storage phosphor screen for 3–5 days.
NOTE: An alternative to screening the dried gel is the transfer of proteins to a nitrocellulose membrane by electro-blotting and screening it thereafter. It results in stronger signals and sharper bands. - Scan the screen on a phosphorimager.
NOTE: As an alternative to phosphor imaging, X-ray film can also be used to reveal the signals.
Representative Results
Following the protocol described above, we assigned mitochondrial translation products from two S. cerevisiae strains: the wild type (WT) and a mutant bearing deletion of the AIM23 gene (AIM23Δ), encoding mitochondrial translation initiation factor 3 (Table 1)8. Mitochondrial translation products were radioactively labeled and separated in SDS-PAAG9. The samples were collected every 2.5 min before saturation to build a time course (Figure 1A). The gel was stained, dried, and screened after the 5-day exposition (Figure 1A).
In the case of a successful experiment, the picture demonstrates eight bands assigned according to the standard pattern4. However, the intensities of individual bands can be highly variable depending on the strain and experimental conditions. Each band corresponds to one translation product. The data (Figure 1A) suggest that the AIM23Δ strain is capable of mitochondrial protein synthesis because all products appearing in the WT are visible in this mutant. However, the intensities of the bands are different from the WT, meaning that the deletion of AIM23 affects mitochondrial gene expression8. Coomassie Brilliant Blue staining serves as a loading control.
The resulting data can be quantified (Figure 1B) to identify differences between strains or experimental conditions using ImageJ10 or ImageQuant software. For this, the ratios of the signal corresponding to every product to the total signal are calculated. Mean values and standard deviations are calculated in at least three independent experiments.
The kinetics of synthesized protein turnover is studied in a pulse-chase experiment (Figure 1C). Samples are collected at the indicated time points after the labeling reaction is stopped by cold methionine and puromycine in step 2.4. This control is necessary to estimate the stability of the products because the intensity of the signal is a result of two opposite processes: synthesis of new chains and protein degradation. Immunostaining with anti-porin 1 antibodies is a loading control.
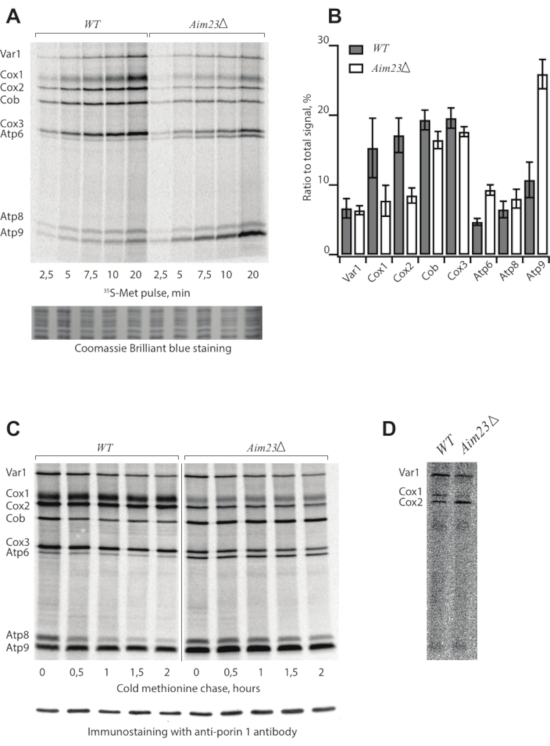
Figure 1: Representative radioactive labeling of yeast mitochondrial translation products. (A) Time course of 35S-methionine incorporation in mitochondrially synthesized proteins in live yeast cells of WT and AIM23Δ strains. Coomassie Brilliant Blue staining is a loading control. (B) Levels of mitochondrially-encoded proteins after 5 min labeling with 35S-methionine. The relative expression is normalized to the total expression of mitochondrially encoded protein genes. Error bars indicate the standard deviation of the mean of at least three independent experiments. (C) Turnover of mitochondrially synthesized proteins in wild type and Aim23Δ strains. The labeling was stopped and the samples were collected at the indicated time points. Immunostaining with anti-porin 1 antibody is a loading control. (D) Sub-optimal experiment with old 35S-methionine, radioautography. Figure 1A,B,C are adapted from8 with minor modifications. Please click here to view a larger version of this figure.
| strain | genotype |
| WT | MATα mal |
| AIM23Δ | MATα mal, AIM23::KanMX4 |
Table 1. Genotypes of S. cerevisiae strains
Discussion
Investigations of gene expression occupy a central part in modern life sciences. Numerous methods providing insights into this complex process have been developed. Here, we described the method allowing to access protein biosynthesis in baker's yeast S. cerevisiae mitochondria. It is usually applied to compare translation efficiencies of the mRNAs in mitochondria of mutant yeast strain versus wild type to access the consequences of the studied mutation. This is one of the basic experiments the researchers conduct when they study the mitochondrial function of yeast cells bearing the mutation suggested to influence mitochondria8,11,12,13. It is often combined with the measurements of oxygen consumption rate and mitochondrial membrane potential. However, the information it provides is not sufficient to distinguish what stage of gene expression is affected. A set of additional experiments is required to find it out. First, northern blot or RT-qPCR evaluation of mitochondrial mRNAs is necessary to assess the transcriptional step. Second, a Western blot of total protein extracts with specific antibodies should be done to assess the protein level. Third, the pulse of labeled 35S-methionine (hot) should be continued with the addition of unlabeled (cold) methionine (chase) and several time points should be collected and analyzed on the gel to investigate the stability of the proteins.
Accurate analysis of mitochondrial gene expression using 35S pulse labeling requires control reactions, especially when the researcher lacks the experience handling it or works with a new yeast strain or a mutant. In these cases, good negative control is a rho0 strain devoid of mitochondrial DNA. It shows efficient cycloheximide inhibition of cytosolic translation and confirms that the banding pattern is mitochondrial translation specific. If the rho0 strain is not available, then we suggest including chloramphenicol along with cycloheximide to inhibit all protein synthesis to confirm cycloheximide efficiency and specificity of the banding pattern.
The closest modification of the protocol is the pulse-chase when the culture is incubated in the shaker (step 2.4) longer than suggested in the pulse experiment (Figure 1C). It is used to study the turnover and stability of mitochondrial translation products. There is another modification of the method when the radioactive labeling is done in organello, not in vivo4. It suggests the isolation of mitochondria from yeast cells. This modification is faster if frozen mitochondria were previously stocked in aliquots. Another advantage is the absence of cycloheximide treatment, which affects different aspects of cellular metabolism. However, isolation of mitochondria and freeze-thawing them can perturb the translation complexes in the organelles providing an artificial picture. Another important modification of the protocol can be done after the separation of mitochondrial translation products in polyacrylamide gel (step 5). Instead of Coomassie Brilliant Blue staining and drying the gel, the protein can be transferred to the nitrocellulose membrane by electro-blotting. This results in stronger and sharper signals. The main reason is that 35S decays by emission of beta particles with very short penetration, so the signal is easily screened in this approach. Electrophoretic conditions can also be modified to provide better resolution. One point is to add 6M urea in the gel, which improves the separation of atp8 and atp914. Another way is using gradient 15%–20% gels.
Translational profiling15 is the method used to dive deeper into the alterations of mitochondrial translation. As compared to radioactive labeling, it allows establishing positions of mitochondrial ribosomes on the mRNA, which makes it possible to find the exact step (initiation, elongation, or termination) being affected. However, profiling is much more expensive, complicated, and time-consuming. Rationally it can be done after the label incorporation experiment, not vice versa. Recently, a novel approach to monitoring yeast mitochondrial translation has been developed16. It avoids treatment with cycloheximide, which is advantageous because such treatment affects cellular metabolism and signaling pathways. Instead of radioactively labeled amino acid incorporation, it utilizes the insertion of a recoded gene for super-folded GFP (sfGFP) in yeast mitochondrial genome, which allows direct measurement of mitochondrial translation using flow cytometry. However, the application of this approach requires the special yeast strain with modified mitochondrial DNA containing sfGFP coding sequence placed between 5’- and 3’- flanking sequences of a certain mitochondrial gene.
The label incorporation experiment includes several critical steps, which cannot be compromised in a successful experiment. First, fresh yeast cultures should be used (step 1.1). Keeping yeasts on the plates longer than 1 month is not recommend; otherwise, they can behave unpredictably in this assay. Second, the cycloheximide solution should be prepared fresh before the experiment and stored frozen no longer than 1 week (step 2.1). The old solution loses its ability to inhibit cytoplasmic translation resulting in a completely aberrant band pattern in the radioautography. Third, 35S-methionine should be fresh and active (step 2.2), otherwise, the intensities of the bands will be weak (Figure 1D). Using the reagent that passed four half-lives (4 x 87.4 days) is not recommended. Avoid boiling the protein samples at 95 °C as standard sample preparation guides suggest (step 4.8) because mitochondrial proteins are highly hydrophobic and prone to aggregation.
There are several common issues one can experience dealing with this method. The first one is the weak intensity of the bands on the radioautography. To fix it make sure, that fresh 35S-methionine is used, a sufficient amount of yeast cells is taken, and the proteins do not aggregate in the pockets on the gel, which can be controlled by Coomassie staining. Keep the dried gel with the screen for no less than 3 days. The second issue is the incorrect band pattern. If it is encountered, make sure that fresh yeast plates and freshly prepared cycloheximide are used. Keep in mind that relative intensities of the bands can vastly vary in different yeast strains and electrophoresis conditions. It makes little sense to load the protein molecular weight ladder on the gel since mitochondrial translation products are never separated according to their molecular masses in this procedure because they are highly hydrophobic. Thus, Cox I (58 kDa) migrates faster than Var 1 (47 kDa). Depending on the buffer conditions, the proteins can even switch positions with one of the neighbors. The third common issue is the blurred picture with no sharp bands observed after Coomassie staining. It indicates the mistakes in the casting of the gel, incorrect buffer composition, or degradation of proteins in the samples. It is recommended to prepare new gel buffers and running buffer carefully checking the composition and the pH values.
Divulgazioni
The authors have nothing to disclose.
Acknowledgements
This research was funded by the Russian Foundation for Basic Research, grant number 18-29-07002. P.K. was supported by State Assignment of Ministry of Science and Higher Education of the Russian Federation, grant number AAAA-A16-116021660073-5. M.V.P. was supported by the Ministry of Science and Higher Education of the Russian Federation, grant number 075-15-2019-1659 (Program of Kurchatov Center of Genome Research). The work was partly done on the equipment purchased in the frame of the Moscow State University Program of Development. I.C., S.L., and M.V.B. were additionally supported by Moscow State University grant “Leading Scientific School Noah’s Ark”.
Materials
| 2-Mercaptoethanol | Sigma-Aldrich | M3148 | |
| Acrylamide | Sigma-Aldrich | A9099 | |
| Ammonium persulfate | Sigma-Aldrich | A3678 | |
| Bacteriological agar | Sigma-Aldrich | A5306 | |
| Biowave Cell Density Meter CO8000 | BIOCHROM US BE | 80-3000-45 | |
| BRAND standard disposable cuvettes | Sigma-Aldrich | Z330361 | |
| chloroform | Sigma-Aldrich | 288306 | |
| cycloheximide | Sigma-Aldrich | C1988 | |
| D-(+)-Galactose | Sigma-Aldrich | G5388 | |
| D-(+)-Glucose | Sigma-Aldrich | G7021 | |
| digital block heater | Thermo Scientific | 88870001 | |
| EasyTag L-[35S]-Methionine, 500µCi (18.5MBq), Stabilized Aqueous Solution | Perkin Elmer | NEG709A500UC | |
| Eppendorf Centrifuge 5425 | Thermo Scientific | 13-864-457 | |
| GE Storage Phosphor Screens | Sigma-Aldrich | GE29-0171-33 | |
| L-methionine | Sigma-Aldrich | M9625 | |
| methanol | Sigma-Aldrich | 34860 | |
| N,N,N′,N′-Tetramethylethylenediamine | Sigma-Aldrich | T9281 | |
| N,N′-Methylenebisacrylamide | Sigma-Aldrich | M7279 | |
| New Brunswick Innova 44/44R Shaker Incubator | New Brunswick Scientific | ||
| Peptone from meat, bacteriological | Millipore | 91249 | |
| Phenylmethanesulfonyl fluoride | Sigma-Aldrich | P7626 | |
| Pierce 660nm Protein Assay Kit | Thermo Scientific | 22662 | |
| PowerPac Basic Power Supply | Bio-Rad | 1645050 | |
| Protean II xi cell | Bio-Rad | 1651802 | |
| Puromycin dihydrochloride from Streptomyces alboniger | Sigma-Aldrich | P8833 | |
| Sodium hydroxide | Sigma-Aldrich | 221465 | |
| Storm 865 phosphor imager | GE Healthcare | ||
| Trizma base | Sigma-Aldrich | 93352 | |
| Vacuum Heated Gel Dryer | Cleaver Scientific | CSL-GDVH | |
| Yeast extract | Sigma-Aldrich | Y1625 |
Riferimenti
- Taylor, R. W., Turnbull, D. M. Mitochondrial DNA mutations in human disease. Nature Reviews. Genetics. 6 (5), 389-402 (2005).
- Park, C. B., Larsson, N. G. Mitochondrial DNA mutations in disease and aging. The Journal of Cell Biology. 193 (5), 809-818 (2011).
- Goffeau, A., et al. Life with 6000 genes. Science. 274 (5287), 546-563 (1996).
- Westermann, B., Herrmann, J. M., Neupert, W. Analysis of mitochondrial translation products in vivo and in organello in yeast. Methods in Cell Biology. 65, 429-438 (2001).
- Foury, F., Roganti, T., Lecrenier, N., Purnelle, B. The complete sequence of the mitochondrial genome of Saccharomyces cerevisiae. FEBS Letters. 440 (3), 325-331 (1998).
- Desai, N., Brown, A., Amunts, A., Ramakrishnan, V. The structure of the yeast mitochondrial ribosome. Science. 355 (6324), 528-531 (2017).
- Sasarman, F., Shoubridge, E. A. Radioactive labeling of mitochondrial translation products in cultured cells. Methods in Molecular Biology. 837, 207-217 (2012).
- Kuzmenko, A., et al. Aim-less translation: loss of Saccharomyces cerevisiae mitochondrial translation initiation factor mIF3/Aim23 leads to unbalanced protein synthesis. Science Reports. 6, 18749 (2016).
- Laemmli, U. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 227 (5259), 680-685 (1970).
- Schneider, C. A., Rasband, W. S., Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nature Methods. 9 (7), 671-675 (2012).
- Keil, M., et al. Oxa1-ribosome complexes coordinate the assembly of cytochrome c oxidase in mitochondria. Journal of Biological Chemistry. 287 (41), 34484-34493 (2012).
- Singhal, R. K., et al. Coi1 is a novel assembly factor of the yeast complex III-complex IV supercomplex. Molecular Biology of the Cell. 28 (20), 2609-2622 (2017).
- Mick, D. U., et al. Coa3 and Cox14 are essential for negative feedback regulation of COX1 translation in mitochondria. The Journal of Cell Biology. 191 (1), 141-154 (2010).
- Bietenhader, M., et al. Experimental relocation of the mitochondrial ATP9 gene to the nucleus reveals forces underlying mitochondrial genome evolution. PLoS Genetics. 8 (8), e1002876 (2012).
- Couvillion, M. T., Churchman, L. S. Mitochondrial ribosome (mitoribosome) profiling for monitoring mitochondrial translation in vivo. Current Protocols in Molecular Biology. 119, 4.28.1-4.28.25 (2017).
- Suhm, T., et al. A novel system to monitor mitochondrial translation in yeast. Microbial Cell. 5 (3), 158-164 (2018).
